
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Messung der Lipolyserate in ex vivo murinem Fettgewebe und primären Präadipozyten, die in vitro differenziert wurden
In diesem Artikel
Zusammenfassung
Die Triglycerid-Lipolyse in Adipozyten ist ein wichtiger Stoffwechselprozess, der zur Freisetzung von freien Fettsäuren und Glycerin führt. Hier stellen wir ein detailliertes Protokoll zur Messung der basalen und stimulierten Lipolyse in Adipozyten und ex vivo Fettgewebe von Mäusen zur Verfügung.
Zusammenfassung
Adipozyten speichern Energie in Form von Triglyceriden in Lipidtröpfchen. Diese Energie kann über die Lipolyse mobilisiert werden, bei der die Fettsäureseitenketten nacheinander vom Glycerinrückgrat abgespalten werden, was zur Freisetzung freier Fettsäuren und Glycerin führt. Aufgrund der geringen Expression der Glycerinkinase in weißen Adipozyten sind die Wiederaufnahmeraten von Glycerin vernachlässigbar, während die Wiederaufnahme von Fettsäuren durch die Fettsäurebindungskapazität von Medienkomponenten wie Albumin bestimmt wird. Sowohl die Freisetzung von Glycerin als auch von Fettsäuren in Medien kann durch kolorimetrische Assays quantifiziert werden, um die lipolytische Rate zu bestimmen. Durch die Messung dieser Faktoren zu mehreren Zeitpunkten kann man die lineare Lipolyserate mit hoher Sicherheit bestimmen. In dieser Arbeit stellen wir ein detailliertes Protokoll für die Messung der Lipolyse in in vitro differenzierten Adipozyten und ex vivo Fettgewebe von Mäusen zur Verfügung. Dieses Protokoll kann auch für andere Präadipozyten-Zelllinien oder Fettgewebe aus anderen Organismen optimiert werden. Überlegungen und Optimierungsparameter werden diskutiert. Dieses Protokoll wurde entwickelt, um die Rate der Adipozyten-Lipolyse zwischen Mausmodellen und Behandlungen zu bestimmen und zu vergleichen.
Einleitung
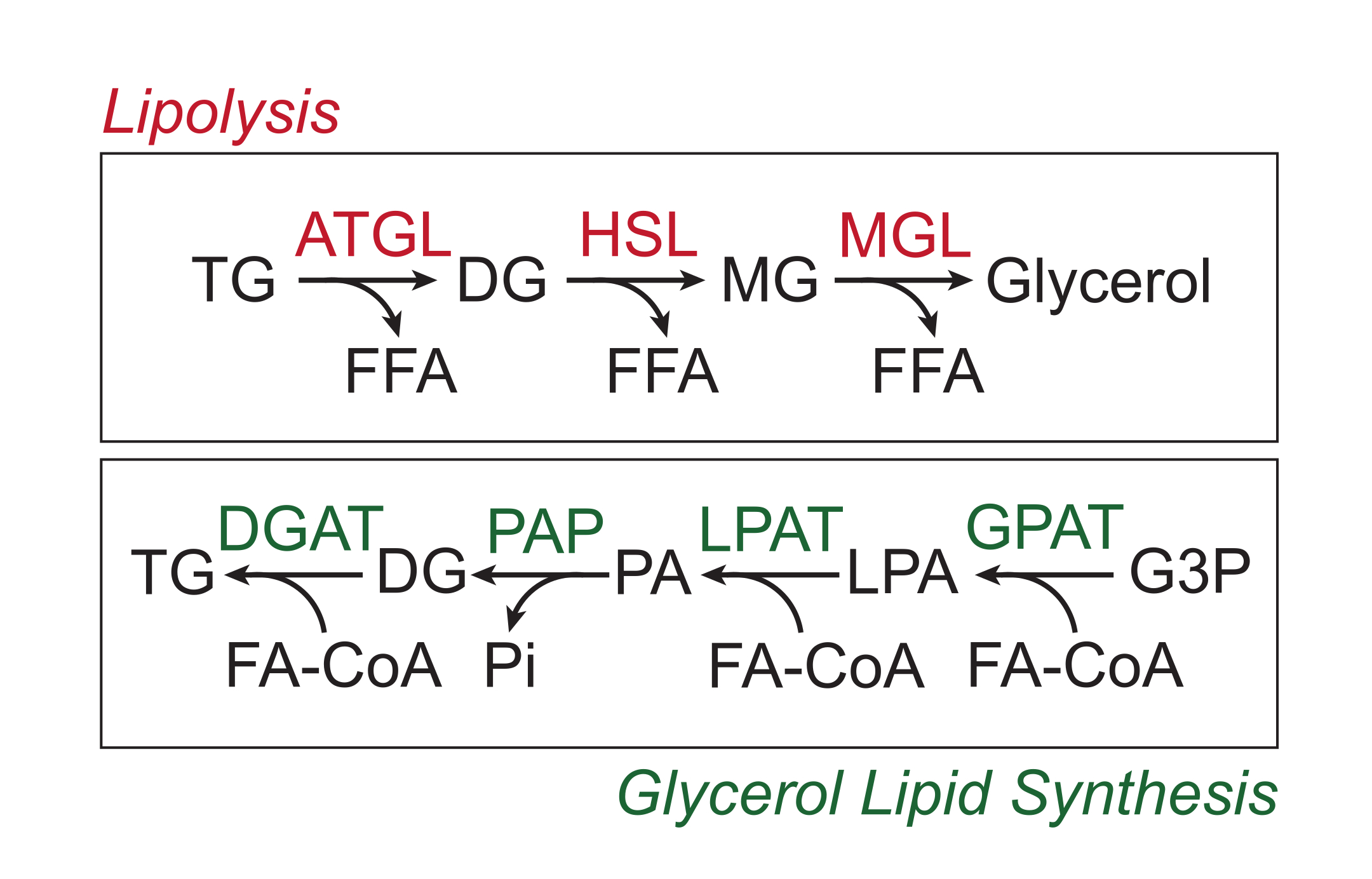
Überschüssige Nährstoffe werden im weißen Fettgewebe in Form von Triglyceriden im neutralen Lipidkern der Lipidtröpfchen gespeichert. Triglyceridspeicher werden über Lipolyse mobilisiert, ein Prozess, bei dem die Fettsäureseitenketten nacheinander durch Fettgewebe-Triglyceridlipase (ATGL), hormonsensitive Lipase (HSL) und Monoglyceridlipase (MGL) gespalten werden, was zur Freisetzung freier Fettsäuren (FFAs) und des Glycerinrückgratsführt 1,2. Die Lipolyse wird durch Katecholamin-Signalübertragung im Fettgewebe aktiviert. Sympathische Nervenendigungen setzen lokal Katecholamine frei, die an β-adrenerge Rezeptoren auf der Plasmamembran der Adipozyten binden. Nach der Ligandenbindung aktivieren diese G-Protein-gekoppelten Rezeptoren (GPCRs) die Adenylylcyclase über Gαs. Die anschließende Aktivierung der Proteinkinase A (PKA) durch cAMP führt zu einer Hochregulation sowohl von ATGL als auch von HSL. Die Phosphorylierung von Perilipin-1 durch PKA bewirkt die Dissoziation von ABHD5 (auch bekannt als CGI-58), das ATGL3 bindet und koaktiviert. PKA phosphoryliert HSL direkt und fördert seine Translokation vom Zytosol zum Lipidtröpfchen, wo die Interaktion mit phosphoryliertem Perilipin-1 die Lipaseaktivität weiter fördert 4,5,6,7. Die dritte Lipase, die an der Lipolyse beteiligt ist, MGL, scheint nicht durch den Katecholamin-Signalweg reguliertzu werden 8. Wichtig ist, dass die Triglyceridsynthese in Adipozyten durch den Glycerinlipidsyntheseweg vermittelt wird, bei dem keine Monoglyceride als Zwischenprodukt gebildet werden. Stattdessen katalysieren Glycerin-3-phosphat-Acyltransferasen die Bildung von Lysophosphatidsäure, die mit einem anderen Fettacyl-CoA zu Phosphatidsäure kombiniert und dann vor der endgültigen Synthese von Triglyceriden zu Diglyceriden isomerisiert wird (Abbildung 1)9,10,11.

Abbildung 1: Lipolyse- und Glycerinlipidsynthesewege. Oben: Lipolytischer Weg; Rot dargestellte Enzyme: Fettgewebe-Triglyceridlipase (ATGL), hormonsensitive Lipase (HSL) und Monoglyceridlipase (MGL). Unten: Glycerin-Lipid-Syntheseweg; Grün dargestellte Enzyme: Diglycerid-Acyltransferase (DGAT), Phosphatidsäure-Phosphatase (PAP), Lysophosphatidsäure-Acyltransferase (LPAT, auch bekannt als LPAATs) und Glycerin-3-Phosphat-Acyltransferase (GPAT). Lipide: Triglyceride (TG), Diglyceride (DG), Monoglyceride (MG), freie Fettsäuren (FFA), Fettacyl-CoA (FA-CoA), Lysophosphatidsäure (LPA) und Phosphatidsäure (PA). Weitere Metaboliten: anorganisches Phosphat (Pi) und Glycerin-3-phosphat (G3P). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Extrazelluläres Adenosin ist ein weiterer wichtiger Regulator der Lipolyse, der durch Gs- und Gi-gekoppelte GPCRs die Aktivität der Adenylcyclase beeinflusst. Der in Adipozyten vorherrschende Adenosinrezeptor ADORA1 hemmt die Adenylylcyclase und damit die Lipolyse durch die Aktivierung von Gi12. ADORA2A wird in niedrigeren Konzentrationen und vor allem in braunen Adipozyten exprimiert und aktiviert die Lipolyse über den Gs-Signalweg 13. ADORA1 beeinflusst sowohl die basale Lipolyse als auch die Reaktion auf adrenerge Agonisten. Die Wirkung von Adenosin auf die Lipolyse kann durch Zugabe von Adenosin-Deaminase zur Neutralisierung von Adenosin sowie des ADORA1-spezifischen Agonisten Phenylisopropyladenosin14,15 kontrolliert werden. Die hormonelle Aktivierung von G q-gekoppelten GPCRs kann auch die Lipolyse über die Aktivierung der Phospholipase C und der Proteinkinase Cbeeinflussen 16,17,18,19. Entzündungssignale wirken sich auch auf die lipolytischen Raten aus. Die Aktivierung von TLR4 durch LPS (und andere Endotoxine) erhöht die lipolytische Rate durch die Aktivierung von ERK, das Perilipin-1 und HSL20 phosphoryliert. TNF-α aktiviert auch die Lipolyse über ERK- und NF-κB-Aktivierung sowie die transkriptionelle Herunterregulierung der Phosphodiesterase PDE-3B und CIDEC21,22,23. IL-6 wurde auch mit einer erhöhten Lipolyse von Adipozyten in Verbindung gebracht, insbesondere im mesenterialen Fettgewebe, dessen FFA-Freisetzung die Lebersteatose und die Gluconeogenese beeinflusst24,25,26.
Die Lipolyse wird im gefütterten Zustand durch Insulin unterdrückt. AKT phosphoryliert und aktiviert PDE-3B, um den cAMP-Signalweg zu unterdrücken und die PKA-Aktivierung zu verhindern27. Insulin reguliert ATGL28 auch transkriptionell herunter. Adipositas fördert die Katecholaminresistenz durch eine Vielzahl von Mechanismen, einschließlich der Herunterregulierung von β-adrenergen Rezeptoren in Adipozyten 29,30,31,32,33. Adipozyten exprimieren alle drei β-adrenergen Rezeptoren (β-1, β-2 und β-3). Während β-1 und β-2 adrenerge Rezeptoren ubiquitär exprimiert werden, wird der β-3 adrenerge Rezeptor in Mäusen überwiegend in Adipozyten exprimiert34,35. Die Expression von Adrb3 wird durch C/EBPα während der Adipogenese induziert36. Der adrenerge Rezeptor β-3 wird in reifen Adipozyten stark exprimiert. Die Aktivierung von β-1 und β-2 adrenergen Rezeptoren ist aufgrund der Rückkopplungshemmung durch β-Arrestin37 selbstlimitierend. Die Hemmung der Rückkopplung des β-3-adrenergen Rezeptors wird durch andere Signalwege vermittelt, die die Adrb3-Expression reduzieren33,38,39.
Zahlreiche Verbindungen können verwendet werden, um die Lipolyse der Adipozyten zu aktivieren. Katecholamine sind wichtige physiologische Aktivatoren der Lipolyse. Noradrenalin (oder Noradrenalin) und Adrenalin (oder Adrenalin) aktivieren alle drei β-adrenergen Rezeptoren40. Noradrenalin und Adrenalin beeinflussen die Lipolyse auch durch die Aktivierung des α-adrenergen Rezeptorsignals41. Häufig verwendete β-adrenerge Rezeptoragonisten sind Isoproterenol, ein nicht-selektiver β-adrenerger Rezeptoragonist, und die β-3-Adrenosensor-Agonisten CL-316,243 und Mirabegron42. Da Adipozyten überwiegend den β-3 adrenergen Rezeptor exprimieren, verwenden wir hier CL-316.243 als Beispiel. Seine Spezifität für den β-3-adrenergen Rezeptor macht es auch zu einem relativ spezifischen Aktivator der Adipozyten-Katecholamin-Signaltransduktion, der auch sicher in vivo verwendet werden kann. Beachten Sie, dass die üblicherweise verwendete Konzentration von 10 μM CL-316,243 in Zellkultur um Größenordnungen höher ist als die ~0,1 μM-Dosis, die erforderlich ist, um ein maximales Ansprechen zu erreichen33. Forskolin umgeht den adrenergen Rezeptor und aktiviert direkt die Adenylylcyclase und die nachgeschaltete lipolytische Signalübertragung. Es gibt noch viele weitere Aktivatoren sowie Unterdrücker der Lipolyse. Bei der Auswahl einer Verbindung zur Stimulierung der Lipolyse sollten die Rezeptorspezifität und die nachgeschalteten Signalwege innerhalb des experimentellen Designs sorgfältig berücksichtigt werden.
Die Lipolyserate im weißen Fettgewebe ist ein wichtiger Stoffwechselfaktor, der sich auf die Kältetoleranz und die Nährstoffverfügbarkeit während des Fastens oder der körperlichen Betätigung auswirkt43,44,45,46. Der Zweck dieses Protokolls ist es, die Lipolyserate in Adipozyten und Fettgewebe zu messen, was das Verständnis des Adipozytenstoffwechsels und dessen Auswirkungen auf den metabolischen Phänotyp verschiedener Mausmodelle erleichtern wird. Um die lipolytische Rate zu quantifizieren, messen wir das Vorkommen lipolytischer Produkte in den Medien (d.h. FFAs und Glycerin). Die Methode beruht auf der Freisetzung lipolytischer Produkte aus den Adipozyten in das Medium. Da weiße Adipozyten geringe Mengen an Glycerinkinase exprimieren, sind die Glycerin-Wiederaufnahmeraten niedrig47. Umgekehrt sollte auch die Produktion von FFAs und Glycerin über andere Stoffwechselwege als die Lipolyse in Betracht gezogen werden. Adipozyten scheinen eine Phosphatase mit Aktivität gegen Glycerin-3-Phosphat zu exprimieren, was die Produktion von Glycerin aus Glycerin-3-phosphat ermöglicht, das aus Glukoseabgeleitet wird 48,49,50. Die Glykolyse ist eine Quelle für Glycerin-3-phosphat, die für die FFA-Wiederveresterung in weißen Adipozyten verwendet wird. Wenn der Glukosespiegel begrenzt ist, benötigt die Glyceroneogenese andere 3-Kohlenstoffquellen wie Laktat undPyruvat 51. Die Kanalisierung von FFAs, die durch Lipolyse innerhalb der Zelle freigesetzt werden, und ihr metabolisches Schicksal sind nur unzureichend verstanden. FFAs, die durch Lipolyse freigesetzt werden, müssen in Fettacyl-CoA umgewandelt werden, bevor sie wieder verestert oder einer β-Oxidation unterzogen werden. Es scheint, dass FFAs, die durch Lipolyse freigesetzt werden, wahrscheinlich die Zelle verlassen, bevor sie wieder aufgenommen und in Fettacyl-CoA umgewandeltwerden 52,53,54,55,56,57,58,59,60,61,62 . FFAs können außerhalb der Zelle durch Albumin sequestriert werden. Wichtig ist, dass langkettige FFAs dafür bekannt sind, die Lipolyse rückkopplungshemmend zu sein, wenn sie nicht durch Albumin 63,64,65,66,67 sequestriert werden. Daher ist die Optimierung der FFA-Pufferkapazität des Mediums während des Lipolyse-Assays von entscheidender Bedeutung. Das hier beschriebene Verfahren ähnelt den bisher veröffentlichten Methoden zur Messung der lipolytischen Rate in Adipozyten und ex vivo Fettgewebe von Mäusen und Menschen 15,68,69,70,71. Dieses Protokoll unterscheidet sich durch die Verwendung von seriellem Sampling; Durch die Durchführung von seriellen Abtastungen können wir intern überprüfen, ob die Lipolyse in der linearen Phase gemessen wird, und mehrere Messungen verwenden, um die Lipolyserate zu berechnen, wodurch Messfehler reduziert werden, um das Vertrauen in den endgültigen berechneten Wert zu erhöhen. Der Nachteil der seriellen Probenahme besteht darin, dass der Assay mehr Zeit und Reagenzien benötigt. Der längere Zeitrahmen verringert jedoch die Auswirkungen von Messfehlern auf den Standardfehler der Schätzungen des Satzes. Darüber hinaus misst dieses Protokoll sowohl die FFA- als auch die Glycerinfreisetzung und berücksichtigt das Verhältnis von FFA:Glycerinfreisetzung mit dem Ziel, ein Verhältnis von 3:1 zu erreichen, wie es bei vollständiger Lipolyse und Freisetzung von lipolytischen Produkten in das Medium zu erwarten wäre72.
Protokoll
Die Verwendung aller Tiere wurde vom Institutional Animal Care and Use Committee (IACUC) am Weill Cornell Medical College der Cornell University genehmigt.
1. Vorbereitung von Puffern und Sammelplatten
- Stellen Sie 5 % Kälberserumalbumin (BSA) her, indem Sie 5 g BSA in 100 ml Dulbeccos modifiziertem Adlermedium (DMEM) ohne Phenolrot auflösen. Rühren Sie das BSA vorsichtig um, damit es sich auflöst (Schütteln ist kontraproduktiv). Sobald das BSA vollständig aufgelöst ist, sterilisieren Sie das Medium mit einem 0,2-μm-Filter. Lagern Sie die BSA-Medien bis zu 1 Monat bei 4 °C.
- Stellen Sie Arbeitskonzentrationen von Kontroll- und Stimulationsmedien her. Steuermedien: 5% BSA-Medien mit Fahrzeugsteuerung. Stimulationsmedien: 5% BSA-Medien mit 0,5 μM CL-316,243. Stellen Sie für jedes Experiment neue Stimulationsmedien her.
- Erwärmen Sie das zu verwendende Medium auf 37 °C. Beschriften Sie eine 96-Well-Platte für die Mediensammlung.
2. Probenvorbereitung
- Führen Sie die Zellkultur wie unten beschrieben durch. Führen Sie alle Zellarbeiten in einem sterilen Abzug durch, um die Kontamination von außen zu minimieren.
- Isolieren und differenzieren Sie primäre Präadipozyten, wie in73,74.
- Platte primäre Präadipozyten mit hoher Dichte, z. B. 1 x 105 Zellen/Well in einer 24-Well-Platte in 1 ml/Well Kulturmedium (15 % fetales Kälberserum (FBS) und 1x Penicillin-Streptomycin-Glutamin in DMEM/F12).
- Nachdem die Zellen eine 100%ige Konfluenz erreicht haben, differenzieren Sie 3 Tage lang mit 5 μM Dexamethason, 0,5 mM 3-Isobutyl-1-methylxanthin, 1 μg/ml Insulin und 1 μM Thiazolidindion (TZD) in Nährmedien. Wechseln Sie dann für mindestens 3 Tage zu Nährmedien mit 1 μg/ml Insulin, um Lipidtröpfchen zu züchten. Verwenden Sie 1 ml/Vertiefung des Mediums in der 24-Well-Platte.
- Wechseln Sie das Nährmedium (1 ml/Well) alle 2 bis 3 Tage mit Insulin. Die Zellen können bis zu 2 Wochen in Medien mit Insulin gehalten werden. Verwenden Sie für diesen Assay nur Kulturen, in denen die Differenzierungsraten über 90 % liegen und in allen Gruppen ähnlich sind, da eine reduzierte Differenzierung als Verringerung der lipolytischen Rate fehlinterpretiert werden könnte.
- Kultivieren Sie die Zellen 24 Stunden lang in insulinfreien Medien, bevor Sie die Lipolyse messen.
HINWEIS: Insulin im Medium hält die Lipidtröpfchen aufrecht, hemmt aber auch die Lipolyse. Die Inkubation ohne Insulin für 24 h ermöglicht eine vollständige lipolytische Aktivierung ohne Verlust des Lipidtröpfchenvolumens. In einigen Systemen muss die Kulturzeit ohne Insulin möglicherweise verkürzt oder verlängert werden.
- Waschen Sie die Zellen einmal mit DPBS, um Serumreste aus dem Nährmedium zu entfernen.
HINWEIS: Dieses Protokoll beinhaltet keinen Serummangel, der die Lipolyse aktivieren kann. Die Serumverhungerung kann nach Ermessen des Forschers eingesetzt werden.
- Isolieren und differenzieren Sie primäre Präadipozyten, wie in73,74.
- Führen Sie die Ex-vivo-Kultur wie unten beschrieben durch.
- Bereiten Sie eine 6-Well-Platte vor, mit einer Vertiefung für jedes Gewebe, das von jeder Maus entnommen werden soll. Geben Sie 4 ml DMEM bei Raumtemperatur in jede zu verwendende Vertiefung.
HINWEIS: BSA in den Sammelmedien ist nicht erforderlich. - Bereiten Sie eine 48-Well-Platte für den Lipolyse-Assay vor, mit einer Well für jedes Replikat. Geben Sie 400 μl DMEM bei Raumtemperatur in jede zu verwendende Vertiefung. Verwenden Sie zwei bis vier Kontroll- und zwei bis vier stimulierte Vertiefungen pro Gewebe und Maus.
- Euthanasieren Sie die Maus durch Zervixluxation unter Narkose mit einer sekundären Methode wie einem bilateralen Pneumothorax. Hier verwendeten wir eine 32 g schwere, 7 Monate alte weibliche C57BL/6J-Maus, die 4 Monate lang mit einer 45% fettreichen Diät gefüttert wurde.
HINWEIS: Dieses Protokoll kann auch für Männer sowie für andere Sorten, Diäten und Altersgruppen verwendet werden. - Mit 70%igem Ethanol besprühen und mit einer Schere einen kleinen (~ 1 cm) seitlichen Schnitt in der Mitte der Bauchhaut machen, die Haut auseinanderziehen, indem Sie mit Daumen und Zeigefinger auf beiden Seiten zusammendrücken und die untere Bauchhaut umklappen, um die hinteren subkutanen Depots freizulegen. Lokalisieren und entfernen Sie den inguinalen Lymphknoten und stumpf präparieren Sie das inguinale Fettgewebe unmittelbar hinter dem inguinalen Lymphknoten mit einer Pinzette.
- Um das Gonadenfettgewebe zu gewinnen, machen Sie einen lateralen und einen vertikalen Schnitt im Bauchfell, um Zugang zur Bauchhöhle zu erhalten. Halten Sie das Gonadenfettpolster mit einer Pinzette fest und schneiden Sie entlang der Gebärmutter (oder des Nebenhodens bei Männern), um das Gonadenfettgewebe zu entfernen. Legen Sie die gesammelten Depots in eine 6-Well-Platte.
- Entfernen Sie das Taschentuch aus der Vertiefung, legen Sie es auf eine Silikonmatte und schneiden Sie es mit einer Schere in 5 bis 7 mg große Stücke.
- Wiegen Sie 25 bis 30 mg (fünf oder sechs Stücke) für jede Assay-Vertiefung ab und geben Sie sie in eine 48-Well-Assay-Platte. Tupfen Sie das Taschentuch vor dem Wiegen auf ein sauberes Handtuch, um alle Medien zu entfernen. Wiegen Sie das Gewichtsboot nach der Entnahme des Gewebes und notieren Sie das Gewicht der zurückgebliebenen Rückstände. Wischen Sie das Gewichtsboot zwischen den Proben sauber und tarieren Sie es bei Bedarf erneut. Verwenden Sie für jedes Gewebe ein neues Gewichtsschiffchen.
- Nachdem alle Gewebeproben gewogen wurden, legen Sie die 48-Well-Assay-Platte für 15 Minuten in einen Inkubator mit 10 % CO2 bei 37 °C und 10 %.
- Bereiten Sie eine 6-Well-Platte vor, mit einer Vertiefung für jedes Gewebe, das von jeder Maus entnommen werden soll. Geben Sie 4 ml DMEM bei Raumtemperatur in jede zu verwendende Vertiefung.
3. Lipolyse-Assay
- Führen Sie die Mediensammlung durch. Führen Sie den Transfer des Mediums und die anschließende Probenentnahme in einem sterilen Abzug durch, um eine mögliche Kontamination durch externe Quellen zu minimieren.
- Entfernen Sie bei t = 0 das Medium und fügen Sie 400 μl pro Well Kontroll- oder Stimulationsmedium hinzu, und stellen Sie die Assay-Platte in einen Inkubator mit 37 °C und 10 % CO2 . Bei Ex-vivo-Gewebekulturen ist das Medium vorsichtig mit einer Pipette zu entfernen. Die Absaugung sollte niemals verwendet werden.
Anmerkungen: Alternativ können Sie eine zweite Platte mit Kontroll- und Stimulationsmedien vorbereiten und das Gewebe übertragen. - Bei t = 1, 2, 3 und 4 h werden 200 μl Medium gesammelt, durch 200 μl des entsprechenden Kontroll- oder Stimulationsmediums ersetzt und die Assayplatte in den Inkubator zurückgelegt. Lagern Sie den Auffangteller bei 4 °C. Um die FFA-Pufferkapazität der BSA-Medien zu ermitteln, verwenden Sie eine zusätzliche Sammlung nach 24 h.
HINWEIS: Die Experimente können hier gestoppt und die gesammelten Medien bei -20 °C gelagert werden.
- Entfernen Sie bei t = 0 das Medium und fügen Sie 400 μl pro Well Kontroll- oder Stimulationsmedium hinzu, und stellen Sie die Assay-Platte in einen Inkubator mit 37 °C und 10 % CO2 . Bei Ex-vivo-Gewebekulturen ist das Medium vorsichtig mit einer Pipette zu entfernen. Die Absaugung sollte niemals verwendet werden.
4. Kolorimetrischer FFA-Assay
- Erwärmen Sie die Reagenzien auf Raumtemperatur und lösen Sie eine Flasche Farbreagenz A mit einer Flasche Lösungsmittel A und eine Flasche Farbreagenz B mit einer Flasche Lösungsmittel B auf. Ab dem Datum der Rekonstitution werden diese Reagenzien am besten innerhalb von 1 Woche verbraucht. 1 Monat nach der Rekonstitution verwerfen.
- Tauen Sie die Proben auf und mischen Sie sie.
- Erstellen Sie eine FFA-Standardkurve. Die Standardlösung ist 1 mM. Verwenden Sie das folgende Volumen mit den Reagenzien für die Standardkurve: 25 μL, 20 μL, 15 μL, 10 μL, 10 μL (1:2 Verdünnung), 10 μL (1:4 Verdünnung), 10 μL (1:8 Verdünnung) und 10 μL Wasser für maximale Reichweite. Für niedrige FFA-Spiegel können 10 μl mit 1 mM, 0,8 mM, 0,6 mM, 0,4 mM, 0,2 mM, 0,1 mM und 0,05 mM Standard besser geeignet sein.
- Pipettieren Sie Standards und Proben in eine 96-Well-Assay-Platte. Das empfohlene Probenvolumen beträgt 10 μl. Fügen Sie drei Vertiefungen mit dem gleichen Volumen an BSA-Medien wie die Proben zur Hintergrundkorrektur hinzu.
Anmerkungen: Wenn die Probenkonzentrationen außerhalb des Bereichs der Standardkurve liegen, wiederholen Sie den Assay und stellen Sie das Probenvolumen auf 2-25 μl ein. - Geben Sie 150 μl Reagenz A in jede Vertiefung und mischen Sie. Vermeiden Sie es, Blasen zu erzeugen. Lass alle Blasen mit einer feinen Nadel platzen. Inkubieren Sie die Assayplatte bei 37 °C für 5 min.
- Lesen Sie die Absorption der Platte bei 550 nm und 660 nm Referenz ab (Messwert A).
- Geben Sie 75 μl Reagenz B in jede Vertiefung und mischen Sie. Vermeiden Sie es, Blasen zu erzeugen. Lass alle Blasen mit einer feinen Nadel platzen. Inkubieren Sie die Assayplatte bei 37 °C für 5 min.
- Lesen Sie die Absorption der Platte erneut bei 550 nm und 660 nm Referenz ab (Messwert B).
5. Kolorimetrischer Glycerin-Assay
- Rekonstituieren Sie das freie Glycerin-Reagenz mit 36 ml Reinstwasser und akklimatisieren Sie es auf Raumtemperatur. Diese Reagenzien werden am besten innerhalb weniger Wochen verwendet. 2 Monate nach der Rekonstitution verwerfen.
- Tauen Sie die Proben auf und mischen Sie sie.
- Erstellen Sie eine Glycerin-Standardkurve, indem Sie eine siebenstufige, 2-fache serielle Verdünnung der Glycerin-Standardlösung und einen Wasserrohling vornehmen.
HINWEIS: Die Standardkurve ist bis zu 25 μl 2,8 mM Glycerin relativ linear, bei höheren Konzentrationen jedoch nicht linear. - Pipettieren Sie jeweils 25 μl Standard und Proben in die 96-Well-Assay-Platte. Fügen Sie drei Vertiefungen mit dem BSA-Medium zur Hintergrundkorrektur hinzu.
- Geben Sie 175 μl freies Glycerinreagenz in jede Vertiefung und mischen Sie. Vermeiden Sie es, Blasen zu erzeugen. Lass alle Blasen mit einer feinen Nadel platzen. Inkubieren Sie die Assayplatte bei 37 °C für 5 min.
- Lesen Sie die Absorption der Platte bei 540 nm ab.
6. Berechnung der lipolytischen Rate
- Beginnen Sie mit den Werten für die optische Dichte (OD). Verwenden Sie für Glycerin direkt die OD-Werte vonA 540 . Berechnen Sie den OD des FFA-Assays nach der folgenden Formel:
OD = (Messwert B: A 550 - A 660) - (Messwert A: A550 - A 660) - Verwenden Sie die Standardkurve, um die FFA- und Glycerinwerte in den gesammelten Proben zu berechnen. Zeichnen Sie die Standard-OD-Werte auf der y-Achse auf und verwenden Sie auf der x-Achse Standardkonzentrationen relativ zum Probenvolumen (d. h. die Konzentration der Wells mit 20 μl 1 mM FFA-Standard auf einer Platte mit 10 μl-Proben entspricht 2 mM). Passen Sie eine lineare Trendlinie an:
y = mx + b - Untersuchen Sie die Standardkurve visuell und entfernen Sie alle Punkte, die außerhalb des linearen Bereichs des Assays liegen. Berechnen Sie die Probenkonzentrationen mit der folgenden Gleichung:
Probenkonzentration: x = (OD - b) ÷ m - Passen Sie Proben an, die außerhalb des linearen Untersuchungsbereichs liegen, und untersuchen Sie sie erneut. Um die endgültige Probenkonzentration zu erhalten, subtrahieren Sie die Konzentration der Hintergrundvertiefungen, die nur BSA-Medien enthalten, von der Konzentration der Proben.
- Berechnen Sie die Mol FFA und Glycerin, die von jeder Probe zu jedem Zeitpunkt produziert werden, gemäß der Formel:

wobei C n = Konzentration zum Zeitpunkt t = n; Vt = Gesamtvolumen im Bohrloch; Vs = Probenentnahmevolumen; und M n = Mol, die zum Zeitpunkt t = n produziert werden (wenn die Konzentrationen in mM und die Volumina in ml angegeben sind, beträgt die Ausgabe μMol).
Zum Beispiel zu verschiedenen Zeitpunkten:
M 1 = C1 × Vt
M 4 = C4 × Vt + (C1 + C2+ C3)Vs
oder
M 4 = C4 × Vt + C3 × V s + C2 × V s + C1 ×V s - Normalisieren Sie das Gewebegewicht, indem Sie es durch das Gewebegewicht für jede Probe in Gramm dividieren, um Einheiten von μmol/g zu erhalten. Für kultivierte Zellen werden die Werte als μmol/well angegeben. Stellen Sie sicher, dass die Zellzahl und die Differenzierungseffizienz von Well zu Well vergleichbar sind.
HINWEIS: Unterschiede in der Proliferations- oder Differenzierungseffizienz erschweren die Interpretation der Ergebnisse und erfordern eine andere Methode der Normalisierung (z. B. Normalisierung auf Protein; siehe Diskussion). - Berechnen Sie die Steigung der erzeugten μmol/g (y-Achse) über der Zeit (x-Achse) für jede Probe einzeln.
- In einer Tabellenkalkulation kann dies mit der Funktion =SLOPE(known_ys,known_xs) erfolgen. Geben Sie in einer neuen Zelle "=SLOPE" ein (markieren Sie dann mit dem Cursor die Glycerin- oder FFA-Werte der Probe in μmol/g und markieren Sie dann die entsprechenden Zeitwerte).
- Überprüfen Sie die Linearität der Daten. R2-Werte sind eine schnelle Möglichkeit, die Linearität der Proben zu bestimmen. In einer Tabellenkalkulation kann dies mit der Funktion =RSQ(known_ys,known_xs) auf die gleiche Weise wie in Schritt 6.7.1 beschrieben erfolgen, aber die anfängliche Eingabe ist =RSQ. Stellen Sie sicher, dass die R2-Werte > 0,98 liegen. Niedrigere Werte weisen auf eine Abweichung von der Linearität hin. Dies kann durch einen Mess-/Abtastfehler oder einen Verlust der Linearität entstehen.
- Eine andere Möglichkeit, die Linearität zu testen, besteht darin, eine lineare Regression für jede Stichprobe durchzuführen und die Residuen darzustellen. Generieren Sie in einer statistischen Analysesoftware eine XY-Tabelle mit einem einzelnen Y-Wert für jeden Zeitpunkt. Wählen Sie Analysieren > Einfache lineare Regression aus, und aktivieren Sie das Kontrollkästchen für Residualdiagramm , bevor Sie auf OK klicken. Das Residuendiagramm wird als neues Diagramm angezeigt.
- Verwenden Sie die FFA- und Glycerinproduktionsrate (d. h. die Steigung [(μmol/g/h]) für jede Probe als individuellen Datenpunkt, um statistische Analysen durchzuführen und Werte darzustellen, wenn verschiedene lipolytische Bedingungen verglichen werden. Wenn lipolytische Raten über Genotypen hinweg verglichen werden, verwenden Sie zwei oder drei Proben pro Tier als technische Replikate und verwenden Sie den Durchschnitt für einen Datenpunkt pro Tier, so dass die Stichprobengröße gleich der Anzahl der Tiere ist.
Ergebnisse
Wir maßen die basale und stimulierte lipolytische Rate von in vitro differenzierten Adipozyten. Primäre Präadipozyten aus inguinalem weißem Fettgewebe wurden durch die Behandlung konfluenter Zellen mit 5 μM Dexamethason, 0,5 mM IBMX, 1 μg/ml Insulin und 1 μM Troglitazon über 4 Tage zu Adipozyten differenziert, gefolgt von einer zusätzlichen 3-tägigen Behandlung mit 1 μg/ml Insulin. Die Zellen wurden vor dem Lipolyse-Assay 24 h lang in Medien ohne Insulin inkubiert. Zum Zeitpunkt = 0h wurden die Zellen...
Diskussion
Hier stellen wir ein grundlegendes Protokoll zur Messung der Lipolyserate in Adipozyten und ex vivo Fettgewebe zur Verfügung. Um die Lipolyse zu quantifizieren, ist es wichtig, die lipolytische Rate in der linearen Phase zu messen. Wir verwenden eine serielle Probenahmetechnik, bei der ein großer Teil der Medien gesammelt und in regelmäßigen Abständen durch frische Medien ersetzt wird. Diese semikonservative Methode ermöglicht die Zugabe von frischem BSA mit FFA-Pufferkapazität und verzögert die Rückkop...
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Diese Arbeit wurde durch das Stipendium der US National Institutes of Health R01DK126944 an S.M.R.
Materialien
| Name | Company | Catalog Number | Comments |
| 24-Well tissue culture treated plate | Corning Inc | 3527 | Must be tissue culture treated for adipocyte differntiation |
| 48-Well flat bottom plate with lid | Corning Inc | 353078 | Can be tissue culture treated |
| 6-Well flat bottom plate with lid | Corning Inc | 353046 | Can be tissue culture treated |
| 96-Well PCR Plate | USA sceintific | 1402-9100 | Any conical 0.2 mL PCR plate will be convenient |
| Bovine Serum Albumin | Sigma Aldrich | A9418 | FFA free BSA such as A8806, is also commonly used. The BSA should not have detectable FFA, also lot to lot variations in BSA can impact the observed rate of lipolysis |
| CL-316,243 | Sigma Aldrich | C5976 | CAS #: 138908-40-4 availaible from other suppliers |
| CO2 incubator | PHCBI | MCO-170AICUVH | CO2 should ideally be set to 10% for adipose tissue, however 5% CO2 will also work |
| DMEM, low glucose, no phenol red | Thermofischer | 11054020 | Any phenol red free media should work, DMEM/F12, RPMI, but should contain volatile buffering capacity, i.e. biocarbonate |
| FFA-free Bovine serum albumin | Equitech-Bio, Inc, | BAH66 | |
| Free Glycerol Reagent | Sigma Aldrich | F6428 | |
| Glycerol Standard Solution | Sigma Aldrich | G7793 | This can also be made by diluting glycerol to the desired concentration |
| HR Series NEFA Standard Solution | Fujifilm | 276-76491 | |
| HR Series NEFA-HR (2) Color Reagent A | Fujifilm | 999-34691 | |
| HR Series NEFA-HR (2) Color Reagent B | Fujifilm | 991-34891 | |
| HR Series NEFA-HR (2) Solvent A | Fujifilm | 995-34791 | |
| HR Series NEFA-HR (2) Solvent B | Fujifilm | 993-35191 | |
| Microbiological Incubator | Fischer Scientific | S28668 | Any incubator at 37C can be used |
| Nunc MicroWell 96-Well Plates | Thermo Scientific | 269620 | Any optically clear, flat bottom 96-well plate works |
| Silicone Laboratory Benchtop Mat | VWR | 76045-300 | Glass plate can also be used. Absorbant surfaces are not recommended |
| Spectrophotometer/Microplate Reader | Molecular devices | SpectraMax i3x | Any plate reader that can read at 540, 550 and 660 mm will work |
| V Bovine serum albumin | Sigma-Aldrich | 810531 | |
| WypAll X70 Wipers | Kimberly-Clark | 41200 | Any high quality paper towel will work |
Referenzen
- Vaughan, M., Berger, J. E., Steinberg, D. Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. The Journal of Biological Chemistry. 239, 401-409 (1964).
- Zimmermann, R., et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 306 (5700), 1383-1386 (2004).
- Lass, A., et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metabolism. 3 (5), 309-319 (2006).
- Stralfors, P., Bjorgell, P., Belfrage, P. Hormonal regulation of hormone-sensitive lipase in intact adipocytes: identification of phosphorylated sites and effects on the phosphorylation by lipolytic hormones and insulin. Proceedings of the National Academy of Sciences. 81 (11), 3317-3321 (1984).
- Miyoshi, H., et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. The Journal of Biological Chemistry. 281 (23), 15837-15844 (2006).
- Sztalryd, C., et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. The Journal of Cell Biology. 161 (6), 1093-1103 (2003).
- Lafontan, M., Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Progress in Lipid Research. 48 (5), 275-297 (2009).
- Grabner, G. F., Xie, H., Schweiger, M., Zechner, R. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nature Metabolism. 3 (11), 1445-1465 (2021).
- Weiss, S. B., Kennedy, E. P., Kiyasu, J. Y. The enzymatic synthesis of triglycerides. The Journal of Biological Chemistry. 235, 40-44 (1960).
- Kennedy, E. P. Biosynthesis of complex lipids. Federation Proceedings. 20, 934-940 (1961).
- Wendel, A. A., Lewin, T. M., Coleman, R. A. Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochimica et Biophysica Acta. 1791 (6), 501-506 (2009).
- Johansson, S. M., Lindgren, E., Yang, J. N., Herling, A. W., Fredholm, B. B. Adenosine A1 receptors regulate lipolysis and lipogenesis in mouse adipose tissue-interactions with insulin. European Journal of Pharmacology. 597 (1-3), 92-101 (2008).
- Gnad, T., et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature. 516 (7531), 395-399 (2014).
- Fried, S. K., et al. Resistance to the antilipolytic effect of insulin in adipocytes of African-American compared to Caucasian postmenopausal women. Journal of Lipid Research. 51 (5), 1193-1200 (2010).
- Lee, M. J., Fried, S. K. Optimal protocol for the differentiation and metabolic analysis of human adipose stromal cells. Methods in Enzymology. 538, 49-65 (2014).
- Fricke, K., Heitland, A., Maronde, E. Cooperative activation of lipolysis by protein kinase A and protein kinase C pathways in 3T3-L1 adipocytes. Endocrinology. 145 (11), 4940-4947 (2004).
- Bergan, H. E., Kittilson, J. D., Sheridan, M. A. PKC and ERK mediate GH-stimulated lipolysis. Journal of Molecular Endocrinology. 51 (2), 213-224 (2013).
- Schmitz-Peiffer, C. The tail wagging the dog--regulation of lipid metabolism by protein kinase C. The FEBS Journal. 280 (21), 5371-5383 (2013).
- Carmen, G. Y., Victor, S. M. Signalling mechanisms regulating lipolysis. Cellular Signalling. 18 (4), 401-408 (2006).
- Zu, L., et al. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. The Journal of Biological Chemistry. 284 (9), 5915-5926 (2009).
- Zhang, H. H., Halbleib, M., Ahmad, F., Manganiello, V. C., Greenberg, A. S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes. 51 (10), 2929-2935 (2002).
- Tan, X., et al. TNF-α downregulates CIDEC via MEK/ERK pathway in human adipocytes. Obesity. 24 (5), 1070-1080 (2016).
- Laurencikiene, J., et al. NF-kappaB is important for TNF-alpha-induced lipolysis in human adipocytes. Journal of Lipid Research. 48 (5), 1069-1077 (2007).
- van Hall, G., et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. The Journal of Clinical Endocrinology and Metabolism. 88 (7), 3005-3010 (2003).
- Wueest, S., et al. Mesenteric fat lipolysis mediates obesity-associated hepatic steatosis and insulin resistance. Diabetes. 65 (1), 140-148 (2016).
- Trujillo, M. E., et al. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. The Journal of Clinical Endocrinology and Metabolism. 89 (11), 5577-5582 (2004).
- Kitamura, T., et al. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Molecular and Cellular Biology. 19 (9), 6286-6296 (1999).
- Chakrabarti, P., et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Molecular and Cellular Biology. 33 (18), 3659-3666 (2013).
- Collins, S., Daniel, K. W., Petro, A. E., Surwit, R. S. Strain-specific response to beta 3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. 138 (1), 405-413 (1997).
- Surwit, R. S., Dixon, T. M., Petro, A. E., Daniel, K. W., Collins, S. Diazoxide restores beta3-adrenergic receptor function in diet-induced obesity and diabetes. Endocrinology. 141 (10), 3630-3637 (2000).
- Gettys, T. W., et al. Age-dependent changes in beta-adrenergic receptor subtypes and adenylyl cyclase activation in adipocytes from Fischer 344 rats. Endocrinology. 136 (5), 2022-2032 (1995).
- Mowers, J., et al. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKε and TBK1. eLife. 2, e01119 (2013).
- Valentine, J. M., et al. β3-Adrenergic receptor downregulation leads to adipocyte catecholamine resistance in obesity. The Journal of Clinical Investigation. 132 (2), e153357 (2022).
- Collins, S., et al. Impaired expression and functional activity of the beta 3- and beta 1-adrenergic receptors in adipose tissue of congenitally obese (C57BL/6J ob/ob) mice. Molecular Endocrinology. 8 (4), 518-527 (1994).
- Collins, S., Surwit, R. S. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Progress in Hormone Research. 56, 309-328 (2001).
- Dixon, T. M., Daniel, K. W., Farmer, S. R., Collins, S. CCAAT/enhancer-binding protein alpha is required for transcription of the beta 3-adrenergic receptor gene during adipogenesis. The Journal of Biological Chemistry. 276 (1), 722-728 (2001).
- Lohse, M. J., Benovic, J. L., Codina, J., Caron, M. G., Lefkowitz, R. J. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 248 (4962), 1547-1550 (1990).
- Nantel, F., et al. The human beta 3-adrenergic receptor is resistant to short term agonist-promoted desensitization. Molecular Pharmacology. 43 (4), 548-555 (1993).
- Liggett, S. B., Freedman, N. J., Schwinn, D. A., Lefkowitz, R. J. Structural basis for receptor subtype-specific regulation revealed by a chimeric beta 3/beta 2-adrenergic receptor. Proceedings of the National Academy of Sciences. 90 (8), 3665-3669 (1993).
- Baker, J. G. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. British Journal of Pharmacology. 160 (5), 1048-1061 (2010).
- Lafontan, M. Inhibition of epinephrine-induced lipolysis in isolated white adipocytes of aging rabbits by increased alpha-adrenergic responsiveness. Journal of Lipid Research. 20 (2), 208-216 (1979).
- Baker, J. G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. British Journal of Pharmacology. 144 (3), 317-322 (2005).
- Jensen, M. D., Nielsen, S. Insulin dose response analysis of free fatty acid kinetics. Metabolism. 56 (1), 68-76 (2007).
- Jensen, M. D., Haymond, M. W., Gerich, J. E., Cryer, P. E., Miles, J. M. Lipolysis during fasting. Decreased suppression by insulin and increased stimulation by epinephrine. The Journal of Clinical Investigation. 79 (1), 207-213 (1987).
- Heckmann, B. L., et al. Defective adipose lipolysis and altered global energy metabolism in mice with adipose overexpression of the lipolytic inhibitor G0/G1 switch gene 2 (G0S2). The Journal of Biological Chemistry. 289 (4), 1905-1916 (2014).
- Shin, H., et al. Lipolysis in brown adipocytes is not essential for cold-induced thermogenesis in mice. Cell Metabolism. 26 (5), 764.e5-777.e5 (2017).
- Treble, D. H., Mayer, J. Glycerolkinase activity in white adipose tissue of obese-hyperglycaemic mice. Nature. 200, 363-364 (1963).
- Possik, E., et al. New mammalian glycerol-3-phosphate phosphatase: role in beta-cell, liver and adipocyte metabolism. Frontiers in Endocrinology. 12, 706607 (2021).
- Romero Mdel, M., Sabater, D., Fernandez-Lopez, J. A., Remesar, X., Alemany, M. Glycerol production from glucose and fructose by 3T3-L1 cells: a mechanism of adipocyte defense from excess substrate. PLoS One. 10 (10), e0139502 (2015).
- Mugabo, Y., et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proceedings of the National Academy of Sciences. 113 (4), E430-E439 (2016).
- Hanson, R. W., Reshef, L. Glyceroneogenesis revisited. Biochimie. 85 (12), 1199-1205 (2003).
- Vaughan, M. The production and release of glycerol by adipose tissue incubated in vitro. The Journal of Biological Chemistry. 237, 3354-3358 (1962).
- Jensen, M. D., Ekberg, K., Landau, B. R. Lipid metabolism during fasting. American Journal of Physiology-Endocrinology and Metabolism. 281 (4), E789-E793 (2001).
- Ballard, F. J., Hanson, R. W., Leveille, G. A. Phosphoenolpyruvate carboxykinase and the synthesis of glyceride-glycerol from pyruvate in adipose tissue. The Journal of Biological Chemistry. 242 (11), 2746-2750 (1967).
- Reshef, L., Hanson, R. W., Ballard, F. J. A possible physiological role for glyceroneogenesis in rat adipose tissue. The Journal of Biological Chemistry. 245 (22), 5979-5984 (1970).
- Gorin, E., Tal-Or, Z., Shafrir, E. Glyceroneogenesis in adipose tissue of fasted, diabetic and triamcinolone treated rats. European Journal of Biochemistry. 8 (3), 370-375 (1969).
- Elia, M., Zed, C., Neale, G., Livesey, G. The energy cost of triglyceride-fatty acid recycling in nonobese subjects after an overnight fast and four days of starvation. Metabolism. 36 (3), 251-255 (1987).
- Reshef, L., et al. Glyceroneogenesis and the triglyceride/fatty acid cycle. Journal of Biological Chemistry. 278 (33), 30413-30416 (2003).
- Edens, N. K., Leibel, R. L., Hirsch, J. Mechanism of free fatty acid re-esterification in human adipocytes in vitro. Journal of Lipid Research. 31 (8), 1423-1431 (1990).
- Vaughan, M., Steinberg, D. Effect of hormones on lipolysis and esterification of free fatty acids during incubation of adipose tissue in vitro. Journal of Lipid Research. 4, 193-199 (1963).
- Brooks, B., Arch, J. R., Newsholme, E. A. Effects of hormones on the rate of the triacylglycerol/fatty acid substrate cycle in adipocytes and epididymal fat pads. Federation of European Biochemical Societies Letters. 146 (2), 327-330 (1982).
- Bjorntorp, P., Karlsson, M., Hovden, A. Quantitative aspects of lipolysis and reesterification in human adipose tissue in vitro. Acta Medica Scandinavica. 185 (1-2), 89-97 (1969).
- Angel, A., Desai, K., Halperin, M. L. Free fatty acid and ATP levels in adipocytes during lipolysis. Metabolism. 20 (1), 87-99 (1971).
- Husted, A. S., et al. Autocrine negative feedback regulation of lipolysis through sensing of NEFAs by FFAR4/GPR120 in WAT. Molecular Metabolism. 42, 101103 (2020).
- Fain, J. N., Shepherd, R. E. Free fatty acids as feedback regulators of adenylate cyclase and cyclic 3':5'-AMP accumulation in rat fat cells. The Journal of Biological Chemistry. 250 (16), 6586-6592 (1975).
- Burns, T. W., Langley, P. E., Terry, B. E., Robinson, G. A. The role of free fatty acids in the regulation of lipolysis by human adipose tissue cells. Metabolism. 27 (12), 1755-1762 (1978).
- Kalderon, B., et al. Suppression of adipose lipolysis by long-chain fatty acid analogs. Journal of Lipid Research. 53 (5), 868-878 (2012).
- Schweiger, M., et al. Measurement of lipolysis. Methods in Enzymology. 538, 171-193 (2014).
- Decaunes, P., Bouloumie, A., Ryden, M., Galitzky, J. Ex vivo analysis of lipolysis in human subcutaneous adipose tissue explants. Bio-Protocol. 8 (3), e2711 (2018).
- Roy, D., Myers, J. M., Tedeschi, A. Protocol for assessing ex vivo lipolysis of murine adipose tissue. STAR Protocols. 3 (3), 101518 (2022).
- Baskaran, P., Thyagarajan, B. Measurement of basal and forskolin-stimulated lipolysis in inguinal adipose fat pads. Journal of Visualized Experiments. 125 (125), 55625 (2017).
- Reilly, S. M., et al. Catecholamines suppress fatty acid re-esterification and increase oxidation in white adipocytes via STAT3. Nature Metabolism. 2 (7), 620-634 (2020).
- Liu, L., et al. Isolation of mouse stromal vascular cells for monolayer culture. Methods in Molecular Biology. 1566, 9-16 (2017).
- DeLuca, J. H., Reilly, S. M. . Methods in Molecular Biology. , (2023).
- Richard, G., Vernon, R. A. C. New Perspectives in Adipose Tissue. Butterworth-Heinemann. , (1985).
- Brito, M. N., Botion, L. M., Brito, N. A., Kettelhut, I. C., Migliorini, R. H. Lipolysis and glycerokinase activity in brown adipose tissue of rat fed a high protein, carbohydrate-free diet. Hormone and Metabolic Research. 26 (1), 51-52 (1994).
- Bertin, R. Glycerokinase activity and lipolysis regulation in brown adipose tissue of cold acclimated rats. Biochimie. 58 (4), 431-434 (1976).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten