
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Mesure du taux de lipolyse dans le tissu adipeux murin ex vivo et les préadipocytes primaires différenciés in vitro
Dans cet article
Résumé
La lipolyse des triglycérides dans les adipocytes est un processus métabolique important entraînant la libération d’acides gras libres et de glycérol. Ici, nous fournissons un protocole détaillé pour mesurer la lipolyse basale et stimulée dans les adipocytes et le tissu adipeux ex vivo de souris.
Résumé
Les adipocytes stockent l’énergie sous forme de triglycérides dans les gouttelettes lipidiques. Cette énergie peut être mobilisée via la lipolyse, où les chaînes latérales d’acides gras sont clivées séquentiellement à partir du squelette glycérol, ce qui entraîne la libération d’acides gras libres et de glycérol. En raison de la faible expression de la glycérol kinase dans les adipocytes blancs, les taux de recapture du glycérol sont négligeables, tandis que la recapture des acides gras est dictée par la capacité de liaison aux acides gras des composants multimédias tels que l’albumine. La libération de glycérol et d’acides gras dans les milieux peut être quantifiée par des tests colorimétriques pour déterminer le taux lipolytique. En mesurant ces facteurs à plusieurs points temporels, on peut déterminer le taux linéaire de lipolyse avec une grande confiance. Ici, nous fournissons un protocole détaillé pour la mesure de la lipolyse dans les adipocytes différenciés in vitro et le tissu adipeux ex vivo de souris. Ce protocole peut également être optimisé pour d’autres lignées cellulaires de préadipocytes ou de tissus adipeux provenant d’autres organismes; Les considérations et les paramètres d’optimisation sont discutés. Ce protocole est conçu pour être utile pour déterminer et comparer le taux de lipolyse adipocytaire entre les modèles murins et les traitements.
Introduction
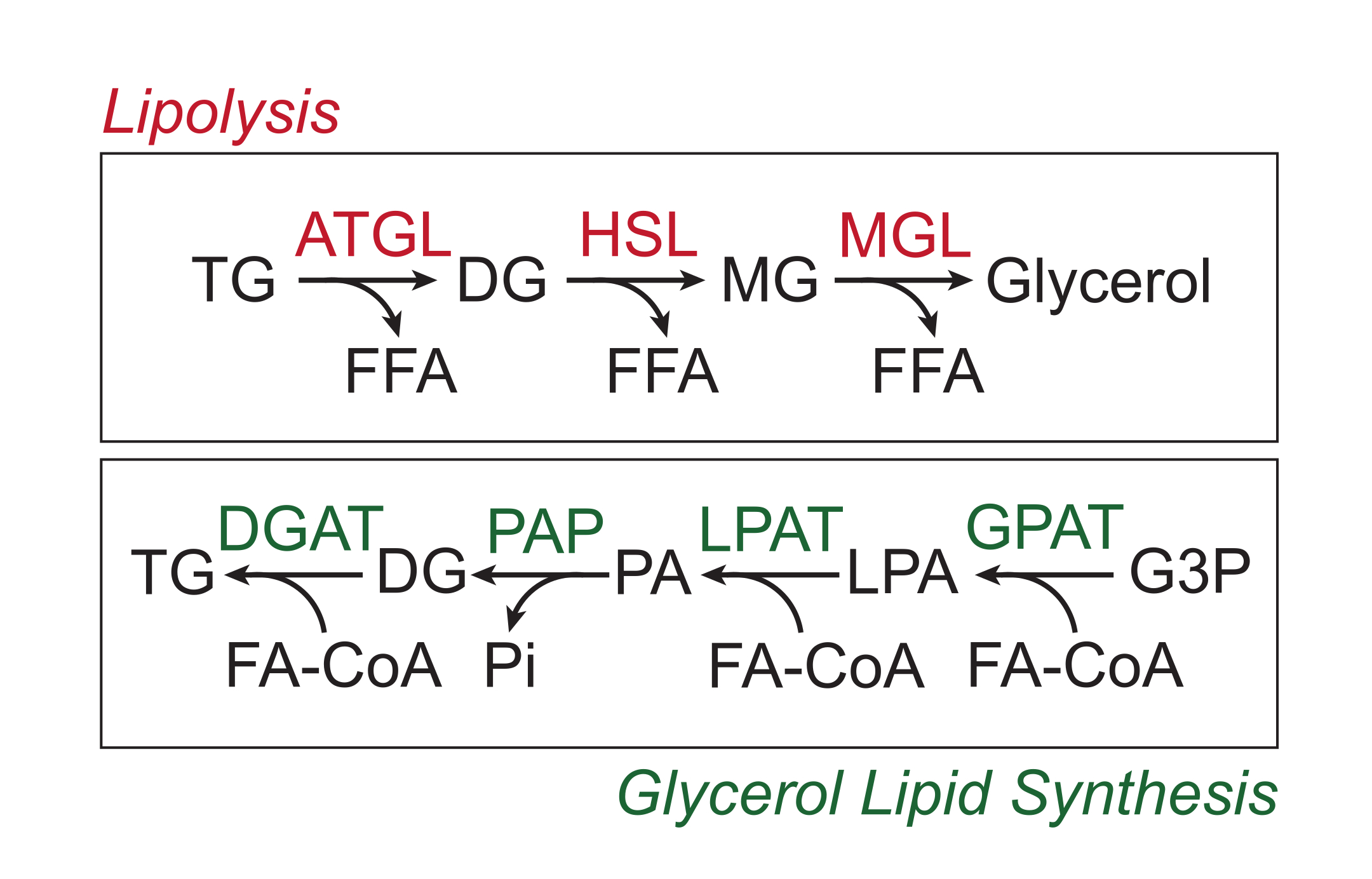
Les nutriments en excès sont stockés dans le tissu adipeux blanc sous forme de triglycérides dans le noyau lipidique neutre des gouttelettes lipidiques. Les réserves de triglycérides sont mobilisées par lipolyse, un processus par lequel les chaînes latérales d’acides gras sont clivées séquentiellement par la triglycéride lipase du tissu adipeux (ATGL), la lipase hormono-sensible (HSL) et la monoglycéride lipase (MGL), ce qui entraîne la libération d’acides gras libres (FFA) et du squelette glycérol 1,2. La lipolyse est activée par la signalisation catécholaminaire dans le tissu adipeux. Les terminaisons nerveuses sympathiques libèrent localement des catécholamines, qui se lient aux récepteurs β-adrénergiques de la membrane plasmique adipocytes. Lors de la liaison au ligand, ces récepteurs couplés aux protéines G (RCPG) activent l’adénylyl cyclase via Gαs. L’activation ultérieure de la protéine kinase A (PKA) par l’AMPc entraîne la régulation positive de l’ATGL et de la HSL. La phosphorylation de la périlipine-1 par la PKA provoque la dissociation d’ABHD5 (également connu sous le nom de CGI-58), qui lie et coactive ATGL3. La PKA phosphoryle directement HSL, favorisant sa translocation du cytosol en gouttelette lipidique, où l’interaction avec la périlipine-1 phosphorylée favorise davantage son activité lipase 4,5,6,7. La troisième lipase impliquée dans la lipolyse, MGL, ne semble pas être régulée par la catécholaminede signalisation 8. Il est important de noter que la synthèse des triglycérides dans les adipocytes est médiée par la voie de synthèse des lipides glycérols, qui n’implique pas la formation de monoglycérides en tant qu’intermédiaire; au lieu de cela, les glycérol-3-phosphate acyl transférases catalysent la formation d’acide lysophosphatidique, qui est combiné avec un autre acyl-CoA gras pour former de l’acide phosphatidique, puis isomérisé en diglycérides avant la synthèse finale des triglycérides (Figure 1)9,10,11.

Figure 1 : Voies de lipolyse et de synthèse des lipides glycérols. En haut : Voie lipolytique ; enzymes indiquées en rouge : triglycérides lipase du tissu adipeux (ATGL), lipase hormonosensible (HSL) et monoglycéride lipase (MGL). En bas: voie de synthèse des lipides glycérol; en vert : diglycéride acyltransférase (DGAT), phosphatidique acide phosphatase (PAP), acyltransférase de l’acide lysophosphatidique (LPAT, également connu sous le nom de LPAAT) et glycérol-3-phosphate acyltransférase (GPAT). Lipides : triglycérides (TG), diglycérides (DG), monoglycérides (MG), acides gras libres (FFA), acyl-CoA gras (FA-CoA), acide lysophosphatidique (LPA) et acide phosphatidique (PA). Autres métabolites : phosphate inorganique (Pi) et glycérol 3-phosphate (G3P). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
L’adénosine extracellulaire est un autre régulateur important de la lipolyse, travaillant à travers les RCPG couplés Gs et Gi pour avoir un impact sur l’activité de l’adényl cyclase. Le récepteur prédominant de l’adénosine dans les adipocytes, ADORA1, inhibe l’adénylylcyclase, et donc la lipolyse par l’activation de Gi12. Exprimé à des niveaux inférieurs, et principalement dans les adipocytesbruns, ADORA2A active la lipolyse via la signalisationG s 13. ADORA1 affecte à la fois la lipolyse basale et la réponse aux agonistes adrénergiques. L’effet de l’adénosine sur la lipolyse peut être contrôlé en ajoutant de l’adénosine désaminase pour neutraliser l’adénosine, ainsi que l’agoniste spécifique ADORA1 phénylisopropyladénosine14,15. L’activation hormonale des RCPG couplés Gq peut également affecter la lipolyse via l’activation de la phospholipase C et de la protéine kinase C16,17,18,19. Les signaux inflammatoires ont également un impact sur les taux lipolytiques. L’activation de TLR4 par le LPS (et d’autres endotoxines) augmente le taux lipolytique en activant ERK, qui phosphoryle la périlipine-1 et la HSL20. Le TNF-α active également la lipolyse via l’activation ERK et NF-κB, ainsi que la régulation transcriptionnelle négative de la phosphodiestérase PDE-3B et CIDEC21,22,23. L’IL-6 a également été associée à une lipolyse adipocytaire accrue, en particulier dans le tissu adipeux mésentérique, dont la libération d’AGL a un impact sur la stéatose hépatique et la gluconéogenèse24,25,26.
La lipolyse est supprimée pendant l’état nourri par l’insuline. AKT phosphoryle et active la PDE-3B pour supprimer la signalisation de l’AMPc et empêcher l’activation de la PKA27. L’insuline régule également transcriptionnellement à la baisse ATGL28. L’obésité favorise la résistance aux catécholamines par divers mécanismes, y compris la régulation négative des récepteurs β-adrénergiques dans les adipocytes 29,30,31,32,33. Les adipocytes expriment les trois récepteurs β-adrénergiques (β-1, β-2 et β-3). Alors que les récepteurs adrénergiques β-1 et β-2 sont omniprésents, le récepteur adrénergique β-3 est principalement exprimé dans les adipocytes chez la souris34,35. L’expression d’Adrb3 est induite par C/EBPα au cours de l’adipogenèse36. Le récepteur adrénergique β-3 est fortement exprimé dans les adipocytes matures. L’activation des récepteurs adrénergiques β-1 et β-2 est spontanément résolutive en raison de l’inhibition de la rétroaction par β-arrestine37. L’inhibition par rétroaction du récepteur adrénergique β-3 est médiée par d’autres voies de signalisation, qui réduisent l’expression d’Adrb3 33,38,39.
De nombreux composés peuvent être utilisés pour activer la lipolyse adipocytes. Les catécholamines sont des activateurs physiologiques majeurs de la lipolyse. La noradrénaline (ou noradrénaline) et l’épinéphrine (ou adrénaline) activent les trois récepteurs β-adrénergiques40. La noradrénaline et l’épinéphrine affectent également la lipolyse via l’activation de la signalisation du récepteur α-adrénergique41. Les agonistes des récepteurs β-adrénergiques couramment utilisés comprennent l’isoprotérénol, qui est un agoniste non sélectif des récepteurs β-adrénergiques, et les agonistes des récepteurs adrénergiques β-3 CL-316,243 et mirabegron42. Étant donné que les adipocytes expriment principalement le récepteur adrénergique β-3, nous utilisons CL-316,243 comme exemple ici. Sa spécificité pour le récepteur adrénergique β-3 en fait également un activateur relativement spécifique de la signalisation des catécholamines adipocytes, qui peut également être utilisé en toute sécurité in vivo. Il est à noter que la concentration couramment utilisée de 10 μM CL-316,243 en culture cellulaire est supérieure de plusieurs ordres de grandeur à la dose de ~0,1 μM requise pour obtenir une réponse maximale33. La forskoline contourne le récepteur adrénergique, activant directement l’adénylyl cyclase et la signalisation lipolytique en aval. Il y a beaucoup plus d’activateurs, ainsi que des suppresseurs de lipolyse. Lors de la sélection d’un composé pour stimuler la lipolyse, la spécificité du récepteur et les voies de signalisation en aval doivent être soigneusement prises en compte dans le plan expérimental.
Le taux de lipolyse dans le tissu adipeux blanc est un facteur métabolique important ayant un impact sur la tolérance au froid et la disponibilité des nutriments pendant le jeûne ou l’exercice43,44,45,46. Le but de ce protocole est de mesurer le taux de lipolyse dans les adipocytes et le tissu adipeux, ce qui facilitera la compréhension du métabolisme des adipocytes et de son impact sur le phénotype métabolique de divers modèles murins. Pour quantifier le taux lipolytique, nous mesurons l’apparition de produits lipolytiques dans les milieux (c.-à-d. AGL et glycérol). La méthode repose sur la libération de produits lipolytiques de l’adipocyte dans le milieu. Étant donné que les adipocytes blancs expriment de faibles niveaux de glycérol kinase, les taux de recapture du glycérol sont faibles47. Inversement, la production d’AGL et de glycérol par des voies métaboliques autres que la lipolyse doit également être envisagée. Les adipocytes semblent exprimer une phosphatase avec une activité contre le phosphate de glycérol-3, permettant la production de glycérol à partir du glycérol-3-phosphate dérivé du glucose48,49,50. La glycolyse est une source de glycérol-3-phosphate utilisée pour la réestérification des AGL dans les adipocytes blancs. Lorsque les niveaux de glucose sont limités, la glycéronéogenèse nécessite d’autres sources de carbone 3, telles que le lactate et le pyruvate51. La canalisation des AGL libérées par la lipolyse au sein de la cellule et leur devenir métabolique sont mal comprises; Les AGL libérés par la lipolyse doivent être convertis en acyl-CoA gras, avant d’être réestérifiés ou de subir une β-oxydation. Il semble que les AGL libérés par la lipolyse sortent probablement de la cellule avant d’être repris et convertis en acyl-CoA gras 52,53,54,55,56,57,58,59,60,61,62 . Les AGL peuvent être séquestrés à l’extérieur de la cellule par l’albumine. Il est important de noter que les AGL à longue chaîne sont connus pour inhiber la lipolyse par rétroaction s’ils ne sont pas séquestrés par l’albumine 63,64,65,66,67. Ainsi, l’optimisation de la capacité tampon FFA du milieu pendant le test de lipolyse est essentielle. La procédure décrite ici est similaire aux méthodes précédemment publiées pour mesurer le taux lipolytique dans les adipocytes et les tissus adipeux ex vivo de souris et d’humains 15,68,69,70,71. Ce protocole diffère par l’utilisation de l’échantillonnage en série; En effectuant un échantillonnage en série, nous pouvons valider en interne que la lipolyse est mesurée dans la phase linéaire et utiliser plusieurs mesures pour calculer le taux de lipolyse, réduisant ainsi l’erreur de mesure pour augmenter la confiance dans la valeur finale calculée. L’inconvénient de l’échantillonnage en série est que le test nécessite plus de temps et de réactifs; Toutefois, la période plus longue réduit l’incidence de l’erreur de mesure sur l’erreur-type des estimations du taux. De plus, ce protocole mesure à la fois la libération de FFA et de glycérol, et considère le rapport de libération de FFA:glycérol dans le but d’atteindre un rapport de 3:1, comme on pourrait s’y attendre d’une lipolyse complète et de la libération de produits lipolytiques dans le milieu72.
Protocole
L’utilisation de tous les animaux a été approuvée par le Comité institutionnel de soin et d’utilisation des animaux (IACUC) du Weill Cornell Medical College de l’Université Cornell.
1. Préparation des tampons et des plaques de collecte
- Obtenir 5 % d’albumine sérique bovine (BSA) en dissolvant 5 g de BSA dans 100 mL de milieu Eagle’s modifié (DMEM) de Dulbecco sans rouge de phénol. Remuez doucement le BSA pour le dissoudre (agiter est contre-productif). Une fois le BSA complètement dissous, stériliser le média avec un filtre de 0,2 μm. Conservez le média BSA à 4 °C jusqu’à 1 mois.
- Faire des concentrations de travail des milieux de contrôle et de stimulation. Média de contrôle: 5% de média BSA avec contrôle du véhicule. Milieux de stimulation : milieu BSA à 5 % avec 0,5 μM CL-316 243. Fabriquez de nouveaux milieux de stimulation pour chaque expérience.
- Réchauffer le média à utiliser à 37 °C. Étiquetez une plaque de 96 puits pour la collecte des médias.
2. Préparation des échantillons
- Effectuer la culture cellulaire comme décrit ci-dessous. Entreprendre tous les travaux cellulaires dans une hotte stérile afin de minimiser la contamination extérieure.
- Isoler et différencier les préadipocytes primaires, comme dans73,74.
- Plaquer les préadipocytes primaires à haute densité, par exemple 1 x 105 cellules/puits dans une plaque de 24 puits dans 1 mL/milieu de culture de puits (15 % de sérum fœtal bovin (FBS) et 1x pénicilline-streptomycine-glutamine dans DMEM/F12).
- Une fois que les cellules ont atteint 100% de confluence, différencier avec 5 μM de dexaméthasone, 0,5 mM de 3-isobutyl-1-méthylxanthine, 1 μg/mL d’insuline et 1 μM de thiazolidinedione (TZD) dans des milieux de culture pendant 3 jours. Ensuite, passez à des milieux de culture avec 1 μg/mL d’insuline pendant au moins 3 jours pour faire croître les gouttelettes lipidiques. Utilisez 1 mL/puits de média dans la plaque de 24 puits.
- Changez le milieu de culture (1 ml/puits) avec de l’insuline tous les 2 ou 3 jours. Les cellules peuvent être maintenues dans des milieux avec de l’insuline jusqu’à 2 semaines. N’utiliser que des cultures dans lesquelles les taux de différenciation sont supérieurs à 90% et sont similaires entre les groupes pour ce test, car une différenciation réduite pourrait être interprétée à tort comme une réduction du taux lipolytique.
- Culture des cellules dans un milieu exempt d’insuline pendant 24 heures avant de mesurer la lipolyse.
REMARQUE: L’insuline dans le milieu maintient les gouttelettes lipidiques, mais inhibe également la lipolyse. L’incubation sans insuline pendant 24 h permet une activation lipolytique complète sans perte de volume de gouttelettes lipidiques. Dans certains systèmes, le temps de culture sans insuline peut devoir être raccourci ou prolongé.
- Lavez les cellules avec DPBS une fois pour éliminer le sérum résiduel du milieu de culture.
REMARQUE: Ce protocole n’inclut pas la famine sérique, qui peut activer la lipolyse. La famine sérique peut être utilisée à la discrétion du chercheur.
- Isoler et différencier les préadipocytes primaires, comme dans73,74.
- Effectuer une culture ex vivo comme décrit ci-dessous.
- Préparez une plaque à 6 puits, avec un puits pour chaque tissu à prélever sur chaque souris. Placez 4 mL de DMEM à température ambiante dans chaque puits à utiliser.
REMARQUE: BSA dans le support de collection n’est pas nécessaire. - Préparer une plaque de 48 puits pour le test de lipolyse, avec un puits pour chaque répétition. Placer 400 μL de DMEM à température ambiante dans chaque puits à utiliser. Utilisez deux à quatre puits témoins et deux à quatre puits stimulés par tissu et par souris.
- Euthanasier la souris par luxation cervicale sous anesthésie, avec une méthode secondaire telle que le pneumothorax bilatéral. Ici, nous avons utilisé une souris C57BL/6J femelle de 32 g, âgée de 7 mois, nourrie avec un régime riche en graisses à 45% pendant 4 mois.
REMARQUE: Ce protocole peut également être utilisé pour les hommes, ainsi que pour d’autres souches, régimes et âges. - Vaporiser avec de l’éthanol à 70% et utiliser des ciseaux pour faire une petite incision latérale (~ 1 cm) au centre de la peau abdominale, écarter la peau en pinçant chaque côté avec le pouce et l’index et plier la peau abdominale inférieure pour révéler les dépôts sous-cutanés postérieurs. Localisez et enlevez le ganglion lymphatique inguinal et disséquez le tissu adipeux inguinal immédiatement postérieur au ganglion lymphatique inguinal à l’aide d’une pince.
- Pour recueillir le tissu adipeux gonadique, faites une incision latérale et verticale dans le péritoine pour accéder à la cavité péritonéale. Tenez le coussinet adipeux gonadique avec une pince à épiler et coupez le long de l’utérus (ou épididyme pour les hommes) pour enlever le tissu adipeux gonadique. Placez les dépôts collectés dans une plaque à 6 puits.
- Retirez le tissu du puits, placez-le sur un tapis de silicone et coupez-le en morceaux de 5 à 7 mg avec des ciseaux.
- Peser 25 à 30 mg (cinq ou six morceaux) pour chaque puits de dosage et placer dans une plaque d’essai de 48 puits. Épongez le mouchoir sur une serviette propre avant de peser pour enlever tout support. Peser le bateau de poids après le retrait du tissu et noter le poids de tout résidu laissé derrière. Essuyez le bateau de poids entre les échantillons et re-tarez si nécessaire. Utilisez un nouveau bateau de musculation pour chaque tissu.
- Une fois que tous les échantillons de tissus ont été pesés, placer la plaque de dosage à 48 puits dans un incubateur à 37 °C, 10% de CO2 pendant 15 min.
- Préparez une plaque à 6 puits, avec un puits pour chaque tissu à prélever sur chaque souris. Placez 4 mL de DMEM à température ambiante dans chaque puits à utiliser.
3. Dosage de la lipolyse
- Effectuer la collecte des médias. Entreprendre le transfert du milieu et le prélèvement subséquent des échantillons dans une hotte stérile afin de réduire au minimum la contamination potentielle provenant de sources extérieures.
- À t = 0, retirer le milieu et ajouter 400 μL par puits de milieu témoin ou de stimulation, puis placer la plaque dans un incubateur à 37 °C, 10 % de CO2 . Pour la culture tissulaire ex vivo , retirer délicatement le milieu à l’aide d’une pipette; L’aspiration ne doit jamais être utilisée.
REMARQUE: Vous pouvez également préparer une deuxième plaque avec des milieux de contrôle et de stimulation et transférer les tissus. - À t = 1, 2, 3 et 4 h, prélever 200 μL de milieu, remplacer par 200 μL du milieu témoin ou de stimulation approprié et remettre la plaque d’essai dans l’incubateur. Conserver la plaque de collecte à 4 °C. Pour déterminer la capacité tampon FFA du média BSA, utilisez une collecte supplémentaire à 24 h.
REMARQUE: Les expériences peuvent être arrêtées ici et les milieux collectés peuvent être stockés à -20 ° C.
- À t = 0, retirer le milieu et ajouter 400 μL par puits de milieu témoin ou de stimulation, puis placer la plaque dans un incubateur à 37 °C, 10 % de CO2 . Pour la culture tissulaire ex vivo , retirer délicatement le milieu à l’aide d’une pipette; L’aspiration ne doit jamais être utilisée.
4. Dosage colorimétrique FFA
- Réchauffer les réactifs à température ambiante et dissoudre un flacon de réactif de couleur A avec un flacon de solvant A et un flacon de réactif de couleur B avec un flacon de solvant B. À partir de la date de reconstitution, il est préférable d’utiliser ces réactifs dans un délai de 1 semaine. Jeter 1 mois après la reconstitution.
- Décongeler et mélanger les échantillons.
- Créez une courbe standard FFA. La solution étalon est de 1 mM. Utilisez le volume suivant avec les réactifs pour la courbe étalon : 25 μL, 20 μL, 15 μL, 10 μL, 10 μL (dilution 1:2), 10 μL (dilution 1:4), 10 μL (dilution 1:8) et 10 μL d’eau pour une plage maximale. Pour les faibles niveaux de FFA, 10 μL de 1 mM, 0,8 mM, 0,6 mM, 0,4 mM, 0,2 mM, 0,1 mM et 0,05 mM standard peuvent être plus applicables.
- Étalons de pipette et échantillons dans une plaque d’essai de 96 puits. Le volume d’échantillon recommandé est de 10 μL. Inclure trois puits avec le même volume de milieux BSA que les échantillons pour la correction de fond.
REMARQUE : Si les concentrations de l’échantillon se situent en dehors de la plage de la courbe standard, répéter l’essai, en ajustant le volume de l’échantillon à 2-25 μL. - Ajouter 150 μL de réactif A à chaque puits et mélanger. Évitez de générer des bulles. Faites éclater les bulles avec une aiguille de calibre fin. Incuber la plaque de dosage à 37 °C pendant 5 min.
- Lire l’absorbance de la plaque à 550 nm et 660 nm de référence (lecture A).
- Ajouter 75 μL de réactif B à chaque puits et mélanger. Évitez de générer des bulles. Faites éclater les bulles avec une aiguille de calibre fin. Incuber la plaque de dosage à 37 °C pendant 5 min.
- Relisez l’absorbance de la plaque à 550 nm et 660 nm de référence (lecture B).
5. Dosage colorimétrique du glycérol
- Reconstituer le réactif glycérol libre avec 36 mL d’eau ultrapure et s’acclimater à température ambiante. Il est préférable d’utiliser ces réactifs en quelques semaines. Jeter 2 mois après la reconstitution.
- Décongeler et mélanger les échantillons.
- Créez une courbe étalon de glycérol en effectuant une dilution en série de sept points, 2 fois, de la solution étalon de glycérol et d’un blanc d’eau.
NOTE: La courbe standard est relativement linéaire jusqu’à 25 μL de glycérol 2,8 mM, mais pas linéaire à des concentrations plus élevées. - Pipeter 25 μL chacun de l’étalon et des échantillons dans la plaque d’essai à 96 puits. Inclure trois puits avec le média BSA pour la correction de fond.
- Ajouter 175 μL de réactif glycérol libre à chaque puits et mélanger. Évitez de générer des bulles. Faites éclater les bulles avec une aiguille de calibre fin. Incuber la plaque de dosage à 37 °C pendant 5 min.
- Lire l’absorbance de la plaque à 540 nm.
6. Calcul du taux lipolytique
- Commencez par les valeurs de densité optique (OD). Pour le glycérol, utilisez directement des valeurs A540 OD. Calculer la DO du dosage FFA selon la formule suivante :
OD = (Lecture B: A 550 - A 660) - (Lecture A: A550 - A 660) - Utilisez la courbe standard pour calculer les niveaux de FFA et de glycérol dans les échantillons prélevés. Tracer les valeurs standard de DO sur l’axe des y, et sur l’axe des x, utiliser les concentrations standard par rapport au volume de l’échantillon (c.-à-d. la concentration des puits avec 20 μL de 1 mM d’AGL standard sur une plaque avec des échantillons de 10 μL est égale à 2 mM). Ajuster une courbe de tendance linéaire :
y = mx + b - Inspectez visuellement la courbe standard et retirez tous les points en dehors de la plage linéaire du test. Calculez les concentrations de l’échantillon à l’aide de l’équation suivante :
Concentration dans l’échantillon: x = (DO - b) ÷ m - Ajuster et réanalyser les échantillons situés en dehors de la plage d’essai linéaire. Pour obtenir la concentration finale de l’échantillon, soustrayez de la concentration des échantillons la concentration des puits de fond ne contenant que des milieux BSA.
- Calculer les moles de FFA et de glycérol produites par chaque échantillon à chaque point temporel, selon la formule:

où C n = concentration au temps t = n ; Vt = volume total dans le puits; Vs = volume de prélèvement de l’échantillon; et M n = moles produites au temps t = n (lorsque les concentrations sont en mM et les volumes en mL, la production est en μMol).
Par exemple, à différents moments :
M 1 = C1 ×V t
M 4 = C4 × Vt + (C1 + C2+ C3)Vs
ou
M 4 = C4 × Vt + C3 × V s + C2 × V s + C1 ×V s - Normaliser le poids des tissus en divisant par le poids tissulaire de chaque échantillon en grammes pour obtenir des unités de μmol/g. Pour les cellules en culture, les valeurs sont présentées en μmol/puits. S’assurer que le nombre de cellules et l’efficacité de différenciation sont comparables d’un puits à l’autre.
REMARQUE : Les différences dans l’efficacité de la prolifération ou de la différenciation compliqueront l’interprétation des résultats et nécessiteront une autre méthode de normalisation (p. ex., normalisation en protéines; voir la discussion). - Calculer la pente du μmol/g produit (axe des y) en fonction du temps (axe des x) pour chaque échantillon individuellement.
- Dans une feuille de calcul, cela peut être fait en utilisant la fonction =SLOPE(known_ys,known_xs). Dans une nouvelle cellule, tapez « =SLOPE » (utilisez ensuite le curseur pour mettre en surbrillance les valeurs de glycérol ou de FFA de l’échantillon en μmol/g, puis pour mettre en surbrillance les valeurs de temps correspondantes).
- Vérifiez la linéarité des données. Les valeurs R2 sont un moyen rapide de déterminer la linéarité des échantillons. Dans une feuille de calcul, cela peut être fait à l’aide de la fonction =RSQ(known_ys,known_xs), de la même manière que décrite à l’étape 6.7.1, mais l’entrée initiale est =RSQ. S’assurer que les valeurs de R2 sont > 0,98; Des valeurs plus faibles indiquent un écart par rapport à la linéarité. Cela peut résulter d’une erreur de mesure/échantillonnage ou d’une perte de linéarité.
- Une autre façon de tester la linéarité consiste à effectuer une régression linéaire pour chaque échantillon et à tracer les résidus. Dans un logiciel d’analyse statistique, générez une table XY avec une seule valeur Y pour chaque point de temps. Sélectionnez Analyser > Régression linéaire simple et cochez la case Tracé résiduel avant d’appuyer sur OK. Le graphique résiduel apparaîtra sous la forme d’un nouveau graphique.
- Utiliser la FFA et le taux de production de glycérol (c.-à-d. pente [(μmol/g/h]) pour chaque échantillon comme point de données individuel pour effectuer une analyse statistique et tracer les valeurs si différentes conditions lipolytiques sont comparées. Si les taux lipolytiques sont comparés entre les génotypes, utiliser deux ou trois échantillons par animal comme répliques techniques et utiliser la moyenne pour un point de données par animal, de sorte que la taille de l’échantillon soit égale au nombre d’animaux.
Résultats
Nous avons mesuré le taux lipolytique basal et stimulé des adipocytes différenciés in vitro. Les préadipocytes primaires du tissu adipeux blanc inguinal ont été différenciés en adipocytes par le traitement de cellules confluentes avec 5 μM de dexaméthasone, 0,5 mM d’IBMX, 1 μg/mL d’insuline et 1 μM de troglitazone pendant 4 jours, suivi d’un traitement supplémentaire de 3 jours avec 1 μg/mL d’insuline. Les cellules ont été incubées dans des milieux sans insuline pendant 24 heures avant ...
Discussion
Ici, nous fournissons un protocole de base pour mesurer le taux de lipolyse dans les adipocytes et le tissu adipeux ex vivo . Pour quantifier la lipolyse, il est important de mesurer le taux lipolytique dans la phase linéaire. Nous utilisons une technique d’échantillonnage en série, où une grande fraction des milieux est collectée et remplacée par des milieux frais à intervalles réguliers. Cette méthode semi-conservatrice permet l’ajout de BSA frais avec une capacité tampon FFA et retarde l’inhib...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce travail a été soutenu par la subvention R01DK126944 des National Institutes of Health des États-Unis à S.M.R.
matériels
| Name | Company | Catalog Number | Comments |
| 24-Well tissue culture treated plate | Corning Inc | 3527 | Must be tissue culture treated for adipocyte differntiation |
| 48-Well flat bottom plate with lid | Corning Inc | 353078 | Can be tissue culture treated |
| 6-Well flat bottom plate with lid | Corning Inc | 353046 | Can be tissue culture treated |
| 96-Well PCR Plate | USA sceintific | 1402-9100 | Any conical 0.2 mL PCR plate will be convenient |
| Bovine Serum Albumin | Sigma Aldrich | A9418 | FFA free BSA such as A8806, is also commonly used. The BSA should not have detectable FFA, also lot to lot variations in BSA can impact the observed rate of lipolysis |
| CL-316,243 | Sigma Aldrich | C5976 | CAS #: 138908-40-4 availaible from other suppliers |
| CO2 incubator | PHCBI | MCO-170AICUVH | CO2 should ideally be set to 10% for adipose tissue, however 5% CO2 will also work |
| DMEM, low glucose, no phenol red | Thermofischer | 11054020 | Any phenol red free media should work, DMEM/F12, RPMI, but should contain volatile buffering capacity, i.e. biocarbonate |
| FFA-free Bovine serum albumin | Equitech-Bio, Inc, | BAH66 | |
| Free Glycerol Reagent | Sigma Aldrich | F6428 | |
| Glycerol Standard Solution | Sigma Aldrich | G7793 | This can also be made by diluting glycerol to the desired concentration |
| HR Series NEFA Standard Solution | Fujifilm | 276-76491 | |
| HR Series NEFA-HR (2) Color Reagent A | Fujifilm | 999-34691 | |
| HR Series NEFA-HR (2) Color Reagent B | Fujifilm | 991-34891 | |
| HR Series NEFA-HR (2) Solvent A | Fujifilm | 995-34791 | |
| HR Series NEFA-HR (2) Solvent B | Fujifilm | 993-35191 | |
| Microbiological Incubator | Fischer Scientific | S28668 | Any incubator at 37C can be used |
| Nunc MicroWell 96-Well Plates | Thermo Scientific | 269620 | Any optically clear, flat bottom 96-well plate works |
| Silicone Laboratory Benchtop Mat | VWR | 76045-300 | Glass plate can also be used. Absorbant surfaces are not recommended |
| Spectrophotometer/Microplate Reader | Molecular devices | SpectraMax i3x | Any plate reader that can read at 540, 550 and 660 mm will work |
| V Bovine serum albumin | Sigma-Aldrich | 810531 | |
| WypAll X70 Wipers | Kimberly-Clark | 41200 | Any high quality paper towel will work |
Références
- Vaughan, M., Berger, J. E., Steinberg, D. Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. The Journal of Biological Chemistry. 239, 401-409 (1964).
- Zimmermann, R., et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 306 (5700), 1383-1386 (2004).
- Lass, A., et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metabolism. 3 (5), 309-319 (2006).
- Stralfors, P., Bjorgell, P., Belfrage, P. Hormonal regulation of hormone-sensitive lipase in intact adipocytes: identification of phosphorylated sites and effects on the phosphorylation by lipolytic hormones and insulin. Proceedings of the National Academy of Sciences. 81 (11), 3317-3321 (1984).
- Miyoshi, H., et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. The Journal of Biological Chemistry. 281 (23), 15837-15844 (2006).
- Sztalryd, C., et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. The Journal of Cell Biology. 161 (6), 1093-1103 (2003).
- Lafontan, M., Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Progress in Lipid Research. 48 (5), 275-297 (2009).
- Grabner, G. F., Xie, H., Schweiger, M., Zechner, R. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nature Metabolism. 3 (11), 1445-1465 (2021).
- Weiss, S. B., Kennedy, E. P., Kiyasu, J. Y. The enzymatic synthesis of triglycerides. The Journal of Biological Chemistry. 235, 40-44 (1960).
- Kennedy, E. P. Biosynthesis of complex lipids. Federation Proceedings. 20, 934-940 (1961).
- Wendel, A. A., Lewin, T. M., Coleman, R. A. Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochimica et Biophysica Acta. 1791 (6), 501-506 (2009).
- Johansson, S. M., Lindgren, E., Yang, J. N., Herling, A. W., Fredholm, B. B. Adenosine A1 receptors regulate lipolysis and lipogenesis in mouse adipose tissue-interactions with insulin. European Journal of Pharmacology. 597 (1-3), 92-101 (2008).
- Gnad, T., et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature. 516 (7531), 395-399 (2014).
- Fried, S. K., et al. Resistance to the antilipolytic effect of insulin in adipocytes of African-American compared to Caucasian postmenopausal women. Journal of Lipid Research. 51 (5), 1193-1200 (2010).
- Lee, M. J., Fried, S. K. Optimal protocol for the differentiation and metabolic analysis of human adipose stromal cells. Methods in Enzymology. 538, 49-65 (2014).
- Fricke, K., Heitland, A., Maronde, E. Cooperative activation of lipolysis by protein kinase A and protein kinase C pathways in 3T3-L1 adipocytes. Endocrinology. 145 (11), 4940-4947 (2004).
- Bergan, H. E., Kittilson, J. D., Sheridan, M. A. PKC and ERK mediate GH-stimulated lipolysis. Journal of Molecular Endocrinology. 51 (2), 213-224 (2013).
- Schmitz-Peiffer, C. The tail wagging the dog--regulation of lipid metabolism by protein kinase C. The FEBS Journal. 280 (21), 5371-5383 (2013).
- Carmen, G. Y., Victor, S. M. Signalling mechanisms regulating lipolysis. Cellular Signalling. 18 (4), 401-408 (2006).
- Zu, L., et al. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. The Journal of Biological Chemistry. 284 (9), 5915-5926 (2009).
- Zhang, H. H., Halbleib, M., Ahmad, F., Manganiello, V. C., Greenberg, A. S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes. 51 (10), 2929-2935 (2002).
- Tan, X., et al. TNF-α downregulates CIDEC via MEK/ERK pathway in human adipocytes. Obesity. 24 (5), 1070-1080 (2016).
- Laurencikiene, J., et al. NF-kappaB is important for TNF-alpha-induced lipolysis in human adipocytes. Journal of Lipid Research. 48 (5), 1069-1077 (2007).
- van Hall, G., et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. The Journal of Clinical Endocrinology and Metabolism. 88 (7), 3005-3010 (2003).
- Wueest, S., et al. Mesenteric fat lipolysis mediates obesity-associated hepatic steatosis and insulin resistance. Diabetes. 65 (1), 140-148 (2016).
- Trujillo, M. E., et al. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. The Journal of Clinical Endocrinology and Metabolism. 89 (11), 5577-5582 (2004).
- Kitamura, T., et al. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Molecular and Cellular Biology. 19 (9), 6286-6296 (1999).
- Chakrabarti, P., et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Molecular and Cellular Biology. 33 (18), 3659-3666 (2013).
- Collins, S., Daniel, K. W., Petro, A. E., Surwit, R. S. Strain-specific response to beta 3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. 138 (1), 405-413 (1997).
- Surwit, R. S., Dixon, T. M., Petro, A. E., Daniel, K. W., Collins, S. Diazoxide restores beta3-adrenergic receptor function in diet-induced obesity and diabetes. Endocrinology. 141 (10), 3630-3637 (2000).
- Gettys, T. W., et al. Age-dependent changes in beta-adrenergic receptor subtypes and adenylyl cyclase activation in adipocytes from Fischer 344 rats. Endocrinology. 136 (5), 2022-2032 (1995).
- Mowers, J., et al. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKε and TBK1. eLife. 2, e01119 (2013).
- Valentine, J. M., et al. β3-Adrenergic receptor downregulation leads to adipocyte catecholamine resistance in obesity. The Journal of Clinical Investigation. 132 (2), e153357 (2022).
- Collins, S., et al. Impaired expression and functional activity of the beta 3- and beta 1-adrenergic receptors in adipose tissue of congenitally obese (C57BL/6J ob/ob) mice. Molecular Endocrinology. 8 (4), 518-527 (1994).
- Collins, S., Surwit, R. S. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Progress in Hormone Research. 56, 309-328 (2001).
- Dixon, T. M., Daniel, K. W., Farmer, S. R., Collins, S. CCAAT/enhancer-binding protein alpha is required for transcription of the beta 3-adrenergic receptor gene during adipogenesis. The Journal of Biological Chemistry. 276 (1), 722-728 (2001).
- Lohse, M. J., Benovic, J. L., Codina, J., Caron, M. G., Lefkowitz, R. J. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 248 (4962), 1547-1550 (1990).
- Nantel, F., et al. The human beta 3-adrenergic receptor is resistant to short term agonist-promoted desensitization. Molecular Pharmacology. 43 (4), 548-555 (1993).
- Liggett, S. B., Freedman, N. J., Schwinn, D. A., Lefkowitz, R. J. Structural basis for receptor subtype-specific regulation revealed by a chimeric beta 3/beta 2-adrenergic receptor. Proceedings of the National Academy of Sciences. 90 (8), 3665-3669 (1993).
- Baker, J. G. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. British Journal of Pharmacology. 160 (5), 1048-1061 (2010).
- Lafontan, M. Inhibition of epinephrine-induced lipolysis in isolated white adipocytes of aging rabbits by increased alpha-adrenergic responsiveness. Journal of Lipid Research. 20 (2), 208-216 (1979).
- Baker, J. G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. British Journal of Pharmacology. 144 (3), 317-322 (2005).
- Jensen, M. D., Nielsen, S. Insulin dose response analysis of free fatty acid kinetics. Metabolism. 56 (1), 68-76 (2007).
- Jensen, M. D., Haymond, M. W., Gerich, J. E., Cryer, P. E., Miles, J. M. Lipolysis during fasting. Decreased suppression by insulin and increased stimulation by epinephrine. The Journal of Clinical Investigation. 79 (1), 207-213 (1987).
- Heckmann, B. L., et al. Defective adipose lipolysis and altered global energy metabolism in mice with adipose overexpression of the lipolytic inhibitor G0/G1 switch gene 2 (G0S2). The Journal of Biological Chemistry. 289 (4), 1905-1916 (2014).
- Shin, H., et al. Lipolysis in brown adipocytes is not essential for cold-induced thermogenesis in mice. Cell Metabolism. 26 (5), 764.e5-777.e5 (2017).
- Treble, D. H., Mayer, J. Glycerolkinase activity in white adipose tissue of obese-hyperglycaemic mice. Nature. 200, 363-364 (1963).
- Possik, E., et al. New mammalian glycerol-3-phosphate phosphatase: role in beta-cell, liver and adipocyte metabolism. Frontiers in Endocrinology. 12, 706607 (2021).
- Romero Mdel, M., Sabater, D., Fernandez-Lopez, J. A., Remesar, X., Alemany, M. Glycerol production from glucose and fructose by 3T3-L1 cells: a mechanism of adipocyte defense from excess substrate. PLoS One. 10 (10), e0139502 (2015).
- Mugabo, Y., et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proceedings of the National Academy of Sciences. 113 (4), E430-E439 (2016).
- Hanson, R. W., Reshef, L. Glyceroneogenesis revisited. Biochimie. 85 (12), 1199-1205 (2003).
- Vaughan, M. The production and release of glycerol by adipose tissue incubated in vitro. The Journal of Biological Chemistry. 237, 3354-3358 (1962).
- Jensen, M. D., Ekberg, K., Landau, B. R. Lipid metabolism during fasting. American Journal of Physiology-Endocrinology and Metabolism. 281 (4), E789-E793 (2001).
- Ballard, F. J., Hanson, R. W., Leveille, G. A. Phosphoenolpyruvate carboxykinase and the synthesis of glyceride-glycerol from pyruvate in adipose tissue. The Journal of Biological Chemistry. 242 (11), 2746-2750 (1967).
- Reshef, L., Hanson, R. W., Ballard, F. J. A possible physiological role for glyceroneogenesis in rat adipose tissue. The Journal of Biological Chemistry. 245 (22), 5979-5984 (1970).
- Gorin, E., Tal-Or, Z., Shafrir, E. Glyceroneogenesis in adipose tissue of fasted, diabetic and triamcinolone treated rats. European Journal of Biochemistry. 8 (3), 370-375 (1969).
- Elia, M., Zed, C., Neale, G., Livesey, G. The energy cost of triglyceride-fatty acid recycling in nonobese subjects after an overnight fast and four days of starvation. Metabolism. 36 (3), 251-255 (1987).
- Reshef, L., et al. Glyceroneogenesis and the triglyceride/fatty acid cycle. Journal of Biological Chemistry. 278 (33), 30413-30416 (2003).
- Edens, N. K., Leibel, R. L., Hirsch, J. Mechanism of free fatty acid re-esterification in human adipocytes in vitro. Journal of Lipid Research. 31 (8), 1423-1431 (1990).
- Vaughan, M., Steinberg, D. Effect of hormones on lipolysis and esterification of free fatty acids during incubation of adipose tissue in vitro. Journal of Lipid Research. 4, 193-199 (1963).
- Brooks, B., Arch, J. R., Newsholme, E. A. Effects of hormones on the rate of the triacylglycerol/fatty acid substrate cycle in adipocytes and epididymal fat pads. Federation of European Biochemical Societies Letters. 146 (2), 327-330 (1982).
- Bjorntorp, P., Karlsson, M., Hovden, A. Quantitative aspects of lipolysis and reesterification in human adipose tissue in vitro. Acta Medica Scandinavica. 185 (1-2), 89-97 (1969).
- Angel, A., Desai, K., Halperin, M. L. Free fatty acid and ATP levels in adipocytes during lipolysis. Metabolism. 20 (1), 87-99 (1971).
- Husted, A. S., et al. Autocrine negative feedback regulation of lipolysis through sensing of NEFAs by FFAR4/GPR120 in WAT. Molecular Metabolism. 42, 101103 (2020).
- Fain, J. N., Shepherd, R. E. Free fatty acids as feedback regulators of adenylate cyclase and cyclic 3':5'-AMP accumulation in rat fat cells. The Journal of Biological Chemistry. 250 (16), 6586-6592 (1975).
- Burns, T. W., Langley, P. E., Terry, B. E., Robinson, G. A. The role of free fatty acids in the regulation of lipolysis by human adipose tissue cells. Metabolism. 27 (12), 1755-1762 (1978).
- Kalderon, B., et al. Suppression of adipose lipolysis by long-chain fatty acid analogs. Journal of Lipid Research. 53 (5), 868-878 (2012).
- Schweiger, M., et al. Measurement of lipolysis. Methods in Enzymology. 538, 171-193 (2014).
- Decaunes, P., Bouloumie, A., Ryden, M., Galitzky, J. Ex vivo analysis of lipolysis in human subcutaneous adipose tissue explants. Bio-Protocol. 8 (3), e2711 (2018).
- Roy, D., Myers, J. M., Tedeschi, A. Protocol for assessing ex vivo lipolysis of murine adipose tissue. STAR Protocols. 3 (3), 101518 (2022).
- Baskaran, P., Thyagarajan, B. Measurement of basal and forskolin-stimulated lipolysis in inguinal adipose fat pads. Journal of Visualized Experiments. 125 (125), 55625 (2017).
- Reilly, S. M., et al. Catecholamines suppress fatty acid re-esterification and increase oxidation in white adipocytes via STAT3. Nature Metabolism. 2 (7), 620-634 (2020).
- Liu, L., et al. Isolation of mouse stromal vascular cells for monolayer culture. Methods in Molecular Biology. 1566, 9-16 (2017).
- DeLuca, J. H., Reilly, S. M. . Methods in Molecular Biology. , (2023).
- Richard, G., Vernon, R. A. C. New Perspectives in Adipose Tissue. Butterworth-Heinemann. , (1985).
- Brito, M. N., Botion, L. M., Brito, N. A., Kettelhut, I. C., Migliorini, R. H. Lipolysis and glycerokinase activity in brown adipose tissue of rat fed a high protein, carbohydrate-free diet. Hormone and Metabolic Research. 26 (1), 51-52 (1994).
- Bertin, R. Glycerokinase activity and lipolysis regulation in brown adipose tissue of cold acclimated rats. Biochimie. 58 (4), 431-434 (1976).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.