
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Vergleichende Studie von Basalmembranmatrizen für die Erhaltung humaner Stammzellen und die Generierung von intestinalen Organoiden
In diesem Artikel
Zusammenfassung
Organoide sind zu wertvollen Werkzeugen für die Modellierung von Krankheiten geworden. Die extrazelluläre Matrix (EZM) steuert das Zellschicksal während der Organoidgenerierung, und die Verwendung eines Systems, das dem nativen Gewebe ähnelt, kann die Modellgenauigkeit verbessern. Diese Studie vergleicht die Erzeugung von induzierten pluripotenten Stammzellen gewonnenen humanen Darmorganoiden in tierischer EZM und xenofreien Hydrogelen.
Zusammenfassung
Die extrazelluläre Matrix (EZM) spielt eine entscheidende Rolle für das Verhalten und die Entwicklung von Zellen. Organoide, die aus humanen induzierten pluripotenten Stammzellen (hiPSCs) erzeugt werden, stehen im Fokus vieler Forschungsbereiche. Der Mangel an physiologischen Hinweisen in klassischen Zellkulturmaterialien behindert jedoch eine effiziente iPSC-Differenzierung. Die Einbeziehung von kommerziell erhältlicher EZM in die Stammzellkultur liefert physikalische und chemische Hinweise, die für die Zellerhaltung von Vorteil sind. Im Handel erhältliche Basalmembranprodukte tierischen Ursprungs bestehen aus EZM-Proteinen und Wachstumsfaktoren, die die Zellerhaltung unterstützen. Da die EZM gewebespezifische Eigenschaften besitzt, die das Zellschicksal modulieren können, werden xenofreie Matrizen verwendet, um die Translation in klinische Studien zu übertragen. Während kommerziell erhältliche Matrizen in der hiPSC- und Organoid-Arbeit weit verbreitet sind, wurde die Äquivalenz dieser Matrizen noch nicht untersucht. In dieser Arbeit wurde eine vergleichende Studie zur hiPSC-Erhaltung und zur Bildung von humanen Darmorganoiden (hIO) in vier verschiedenen Matrices durchgeführt: Matrigel (Matrix 1-AB), Geltrex (Matrix 2-AB), Cultrex (Matrix 3-AB) und VitroGel (Matrix 4-XF). Obwohl die Kolonien keine perfekt runde Form aufwiesen, kam es zu einer minimalen spontanen Differenzierung, wobei über 85% der Zellen den Stammzellmarker SSEA-4 exprimierten. Matrix 4-XF führte zur Bildung von runden 3D-Klumpen. Außerdem verbesserte die Erhöhung der Konzentration von Ergänzungs- und Wachstumsfaktoren in den Medien, die zur Herstellung der Matrix 4-XF-Hydrogellösung verwendet wurden, die hiPSC-Expression von SSEA-4 um das 1,3-fache. Die Differenzierung von Matrix-2-AB-erhaltenen hiPS-Zellen führte zu weniger Sphäroidfreisetzungen im Mittel-/Hinterdarmstadium im Vergleich zu den anderen tierischen Basalmembranen. Im Vergleich zu anderen führt die xenofreie Organoidmatrix (Matrix 4-O3) zu einem größeren und reiferen hIO, was darauf hindeutet, dass die physikalischen Eigenschaften von xenofreien Hydrogelen genutzt werden können, um die Organoidbildung zu optimieren. Insgesamt deuten die Ergebnisse darauf hin, dass Variationen in der Zusammensetzung verschiedener Matrices die Stadien der IO-Differenzierung beeinflussen. Diese Studie schärft das Bewusstsein für die Unterschiede in kommerziell verfügbaren Matrizen und bietet einen Leitfaden für die Matrixoptimierung während der iPSC- und IO-Arbeit.
Einleitung
Die extrazelluläre Matrix (EZM) ist ein dynamischer und multifunktionaler Bestandteil von Geweben, der eine zentrale Rolle bei der Regulierung des Zellverhaltens und der Zellentwicklung spielt. Als komplexes Netzwerk bietet es strukturelle Unterstützung, zellhaftende Liganden1 und die Speicherung von Wachstumsfaktoren und Zytokinen, die die Zellsignalübertragung regulieren. So dient die EZM bei der Wundheilung als Gerüst für wandernde Zellen und als Reservoir für Wachstumsfaktoren, die an der Gewebereparatur beteiligt sind2. In ähnlicher Weise kann eine Dysregulation in der EZM zu einer Zunahme des Schweregrads verschiedener Krankheiten wie Fibrose und Krebs führen 3,4. Während der Embryonalentwicklung steuert die EZM die Morphogenese des Gewebes. Bei der Entwicklung des Herzens spielen beispielsweise ECM-Komponenten eine Rolle bei der Schaffung der korrekten Architektur und Funktion des Herzgewebes5. Über ein Jahrzehnt der Forschung hat gezeigt, dass allein die Steifigkeit der Mikroumgebung 6,7 die Spezifikation der Stammzelllinie steuern kann. Daher ist es nicht verwunderlich, dass die EZM bei der in vitro Zelldifferenzierung das Schicksal der Stammzellen beeinflusst, indem sie Signale zur Differenzierung liefert.
Organoide können aus induzierten pluripotenten Stammzellen (iPSCs) erzeugt werden. Um Organoide erfolgreich zu erzeugen, ist es erforderlich, mit einer richtig charakterisierten iPS-Linie zu beginnen. Der Mangel an physiologischen Hinweisen in klassischen Zellkulturmaterialien behindert jedoch eine effiziente iPSC-Differenzierung und Organoid-Generierung. Darüber hinaus hat die neuere Forschung die Bedeutung der Zusammensetzung der extrazellulären Matrix (EZM), der Wechselwirkungen zwischen Zellen und der EZM8 sowie mechanischer und geometrischer Signale 9,10,11 im Zusammenhang mit der Expansion und Differenzierung von Organoidenhervorgehoben 12. Die Weiterentwicklung der Organoid-Technologie durch Verbesserung der Reproduzierbarkeit erfordert die Einbeziehung gewebespezifischer physikalischer und chemischer Signale.
Organoide zielen darauf ab, das native Gewebe in einer physiologisch ähnlichen Mikroumgebung zu rekapitulieren. Die Wahl eines EZM-Systems, das die EZM des nativen Gewebes genau nachahmt, ist entscheidend für das Erreichen physiologischer Relevanz in Bezug auf das Verhalten, die Funktion und die Reaktion auf Reizevon Zellen 13. Die Wahl der EZM-Komponenten kann die Differenzierung von Stammzellen in bestimmte Zelltypen innerhalb des Organoids beeinflussen. Verschiedene EZM-Proteine und ihre Kombinationen können Hinweise geben, die das Schicksal der Zellen lenken14. So haben Studien gezeigt, dass die Verwendung spezifischer EZM-Komponenten die Differenzierung von Darmstammzellen in reife Darmzelltypen fördern kann, was zu physiologisch relevanten Darmorganoiden führt15. Während Organoide ein wertvolles Werkzeug bei der Modellierung von Krankheiten und bei der Erprobung von Medikamenten sind, ist die Auswahl eines geeigneten ECM-Systems für diese Anwendung von entscheidender Bedeutung. Ein geeignetes EZM-System kann die Genauigkeit der Krankheitsmodellierung verbessern, indem es eine Mikroumgebung schafft, die dem betroffenen Gewebe ähnelt16. Darüber hinaus kann gewebespezifische EZM dazu beitragen, Organoide zu erzeugen, die krankheitsassoziierte Phänotypen und Arzneimittelreaktionen besser rekapitulieren17. Die Optimierung des ECM-Systems, das bei der Organoid-Differenzierung verwendet wird, ist entscheidend für das Erreichen der gewünschten Differenzierungsergebnisse.
Kommerziell erhältliche Basalmembransysteme, die aus tierischen EZM-Quellen gewonnen werden (z. B. Matrigel, Cultrex) und xenofreies Hydrogel (z. B. VitroGel), werden häufig in der iPS-Forschung und Organoidforschung eingesetzt. Unternehmen, die sie vermarkten, und Forscher, die sie verwenden, haben im Laufe der Jahre viele Anleitungen für ihre spezifischen Produkte und Anwendungen erstellt. Viele dieser Anweisungen dienten als Leitfaden für die Erstellung dieses Protokolls. Darüber hinaus wurden die Vor- und Nachteile, die mit ihren intrinsischen Eigenschaften verbunden sind, von vielen einzeln festgestellt 18,19,20,21. Es gibt jedoch keinen systematischen Arbeitsablauf, der die Auswahl optimaler Systeme für die Arbeit mit iPSCs und Organoiden leitet. Hier wird ein Workflow zur systematischen Bewertung der Äquivalenz von ECM-Systemen aus verschiedenen Quellen für iPSC- und Organoid-Arbeit bereitgestellt. Dabei handelt es sich um eine vergleichende Studie zur Aufrechterhaltung der Bildung von zwei verschiedenen humanen iPSC-Linien (hiPSC) und humanen Darmorganoiden (hIO) in vier verschiedenen Matrices: Matrigel (Matrix 1-AB), Geltrex (Matrix 2-AB), Cultrex (Matrix 3-AB) und VitroGel (Matrix 4-XF). Für die Organoidkultur wurden vier Versionen der xenofreien Matrix VitroGel verwendet, die zuvor für die Organoidkultur optimiert waren: ORGANOID 1 (Matrix 4-O1), ORGANOID 2 (Matrix 4-O2), ORGANOID 3 (Matrix 4-O3), ORGANOID 4 (Matrix 4-O4). Außerdem wurden für Organoide optimierte Matrizen tierischen Ursprungs verwendet: Matrigel High Concentration (Matrix 1-ABO) und Cultrex Type 2 (Matrix 3-ABO). Es wurden kommerziell erhältliche Stammzellkulturmedien (mTeSR Plus) und ein Organoid-Differenzierungskit (STEMdiff Darm-Organoid-Kit) verwendet. Dieses Protokoll kombiniert die individuellen Anweisungen der Produkthersteller mit Laborerfahrungen, um den Leser zu einer erfolgreichen Optimierung der ECM für seine spezifische iPSC- und Organoid-Arbeit zu führen. Insgesamt unterstreichen dieses Protokoll und die repräsentativen Ergebnisse die Bedeutung der Auswahl der optimalen Mikroumgebung für die Stammzellarbeit und die Differenzierung von Organoiden.
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. hiPSC-Wartung
ACHTUNG: Alle Arbeiten werden in einer Biosicherheitswerkbank (BSC) nach aseptischen Standardtechniken durchgeführt. Muss die OSHA-Sicherheitsstandards für Labore einhalten, einschließlich der ordnungsgemäßen Verwendung von persönlicher Schutzausrüstung wie Laborkitteln, Handschuhen und Schutzbrillen.
- Vorbereitung von Matrices, Aliquoten und Zellkulturmedien
- Für kommerziell erhältliche Basalmembranen tierischen Ursprungs (BMs; Matrix 1-AB, Matrix 2-AB, Matrix 3-AB), Aliquots der Arbeitsvolumina gemäß der in Tabelle 1 zusammengefassten Empfehlung des Herstellers vorbereiten und für die Langzeitlagerung bei -20 °C oder -80 °C lagern. Vermeiden Sie die Bildung von Blasen. Wenn sich einige Blasen bilden, zentrifugieren Sie die Proben vor der Lagerung bei 4 °C bei <200 x g für ~1-2 Minuten , um die Blase an die Oberfläche zu zwingen.
HINWEIS: Die Herstellung von Einweg-Aliquoten hilft, wiederholte Einfrier-Tau-Zyklen zu vermeiden, die die ECM-Architektur stören. Da die BM-Konzentration mit der Chargennummer variiert, sollten Sie die Empfehlung des Herstellers befolgen, Einmal-Aliquots herzustellen und Beschichtungslösungen herzustellen. Hochkonzentrierte BMs sind viskos und schwer zu pipettieren; Verwenden Sie kalte Spitzen, die zuvor bei -20 °C gelagert wurden. - Bereiten Sie das Stammzellkulturmedium gemäß den Empfehlungen des Herstellers vor. Zur Herstellung des spezifischen kommerziell erhältlichen vollständigen Mediums, das in diesem Protokoll verwendet wird, fügen Sie 100 ml eines 5-fach-Ergänzungscocktails zu 400 ml Basalmedium hinzu. Mischen Sie dann dieses Medium gründlich und aliquotieren Sie es in 40-ml-Volumina, die bei 20 °C gelagert werden. Für den Gebrauch jedes Aliquot des vollständigen Mediums auftauen, das aliquotierte vollständige Medium sofort verwenden oder bis zu 2 Wochen bei 2 - 8 °C lagern. Frieren Sie es nicht wieder ein.
- Zur Herstellung des Matrix 4-XE-Hydrogels wird ein Aliquote des Stammzell-Komplettmediums hergestellt, das die 3-fache Konzentration des Ergänzungscocktails enthält, d.h. das 5-fache der Ergänzung auf das 3-fache verdünnen.
- Für kommerziell erhältliche Basalmembranen tierischen Ursprungs (BMs; Matrix 1-AB, Matrix 2-AB, Matrix 3-AB), Aliquots der Arbeitsvolumina gemäß der in Tabelle 1 zusammengefassten Empfehlung des Herstellers vorbereiten und für die Langzeitlagerung bei -20 °C oder -80 °C lagern. Vermeiden Sie die Bildung von Blasen. Wenn sich einige Blasen bilden, zentrifugieren Sie die Proben vor der Lagerung bei 4 °C bei <200 x g für ~1-2 Minuten , um die Blase an die Oberfläche zu zwingen.
- Beschichtung von Gewebekulturkunststoff mit tierischen BMs (Matrix 1-3AB)
- Bereiten Sie Folgendes vor, bevor Sie beginnen: Bewahren Sie die Matrizen beim Auftauen und bei der Handhabung immer im Eis auf, um ein Gelieren zu verhindern. Verwenden Sie kalte Medien, um die verdünnten Matrizen vorzubereiten. Bereiten Sie genügend Replikationswells pro Bedingung vor, um Zellen vor Beginn und in jeder Phase des hiPSC-Differenzierungsprozesses und der hIO-Organoidgenerierung zu testen.
HINWEIS: Hier wurden für diese Studie 24-Well-Platten verwendet. In Tabelle 2 finden Sie die empfohlenen Beschichtungsvolumina für andere Plattengrößen. - Für jeden Matrixtyp sind 25 ml Kaltvorschub DMEM/F-12 mit 15 mM HEPES in einem konischen 50-ml-Röhrchen vorzubereiten. Bewahren Sie diese auf Eis auf.
- Die aufgetauten Matrizen in ihren jeweiligen Kaltvorlauf DMEM/F-12 geben und gut vermischen. Halten Sie das Medium während des Mischvorgangs kalt. Prüfen Sie visuell auf eine homogene Durchmischung, indem Sie sicherstellen, dass keine Klumpen vorhanden sind.
- Verwenden Sie sofort die verdünnten Matrices-Lösungen, um jede für die Verwendung ausgewählte Vertiefung zu beschichten (250 μl/Vertiefung bei Verwendung von 24-Well-Platten).
- Kippen Sie das Kulturgefäß vorsichtig, um die Beschichtungslösung gleichmäßig auf der Oberfläche zu verteilen. Vor Gebrauch mindestens 1 h bei Raumtemperatur (15 - 25 °C) inkubieren.
HINWEIS: Wenn die Platte nicht sofort verwendet wird, kann sie bis zu 1 Woche nach der Beschichtung bei 2 - 8 °C gelagert werden, muss jedoch mit einer transparenten Folie versiegelt werden, um eine Verdunstung zu verhindern. Wenn Sie gelagerte Platten verwenden, lassen Sie sie 30 Minuten lang Raumtemperatur (15 - 25 °C) erreichen, bevor Sie mit dem nächsten Schritt fortfahren. - Entfernen Sie die überschüssige Lösung vorsichtig mit einer serologischen Pipette oder durch Aspiration. Achten Sie darauf, dass die beschichtete Oberfläche nicht zerkratzt wird.
- Fügen Sie sofort warmes, vollständiges Stammzellmedium hinzu (50 % des Gesamtvolumens, das für die spezifische Vertiefung benötigt wird, z. B. 250 μl/Vertiefung bei Verwendung einer 24-Well-Platte).
- Bereiten Sie Folgendes vor, bevor Sie beginnen: Bewahren Sie die Matrizen beim Auftauen und bei der Handhabung immer im Eis auf, um ein Gelieren zu verhindern. Verwenden Sie kalte Medien, um die verdünnten Matrizen vorzubereiten. Bereiten Sie genügend Replikationswells pro Bedingung vor, um Zellen vor Beginn und in jeder Phase des hiPSC-Differenzierungsprozesses und der hIO-Organoidgenerierung zu testen.
- Beschichtung von Gewebekulturkunststoff mit Matrix 4-XE Hydrogel
- Bereiten Sie vor dem Start folgendes vor: Nehmen Sie das Hydrogel Matrix 4-XE aus dem Kühlschrank und lassen Sie es auf Raumtemperatur (25 °C) erwärmen. Bereiten Sie genügend Replikationswells pro Bedingung vor, um Zellen vor Beginn und in jeder Phase des hiPSC-Differenzierungsprozesses und der hIO-Organoidgenerierung zu testen.
HINWEIS: Hier wurden für diese Studie 24-Well-Platten verwendet. In Tabelle 3 finden Sie die empfohlenen Beschichtungsvolumina für andere Plattengrößen. - Bereiten Sie ein vollständiges Stammzellkulturmedium vor, das die 3-fache Konzentration des Wachstumsfaktors im Vergleich zur Standardformulierung für die Stammzellkultur (3x Stammzellmedium) enthält.
- Mischen Sie Matrix 4-XE Hydrogel und 3x Stammzellmedium in einem Mischungsverhältnis von 2:1 v/v und pipettieren Sie vorsichtig 5x-10x auf und ab, um gründlich zu mischen.
- Übertragen Sie die Hydrogelmischung auf eine Vertiefungsplatte und kippen Sie das Kulturgefäß vorsichtig, um die Mischung gleichmäßig auf der Oberfläche zu verteilen. Verwenden Sie 250 μl/Well, wenn Sie eine 24-Well-Platte verwenden. In Tabelle 3 finden Sie die empfohlenen Volumina.
- Warten Sie 10-15 Minuten bei Raumtemperatur, bis sich ein weiches Gel gebildet hat. Zerstören Sie das Hydrogel während des Hydrogelbildungsprozesses nicht durch Kippen oder Schütteln der Platte.
- Bereiten Sie vor dem Start folgendes vor: Nehmen Sie das Hydrogel Matrix 4-XE aus dem Kühlschrank und lassen Sie es auf Raumtemperatur (25 °C) erwärmen. Bereiten Sie genügend Replikationswells pro Bedingung vor, um Zellen vor Beginn und in jeder Phase des hiPSC-Differenzierungsprozesses und der hIO-Organoidgenerierung zu testen.
- hiPSC-enzymfreie Klumpenpassage und Aussaat
- Bereiten Sie vor dem Start folgendes vor: Mindestens 1 h vor dem Passieren die gewünschten Kunststoffartikel mit Matrizen bestreichen. Aliquotieren Sie ausreichend vollständige Stammzellmedien und erwärmen Sie sie auf Raumtemperatur (15 - 25 °C). Vermeiden Sie mehrfache Heizzyklen für das gesamte Medium.
HINWEIS: Die folgenden Schritte beschreiben die Passage einer bereits etablierten und zu >90 % konfluenten Kultur von iPSCs auf einer 6-Well-Platte und deren Aussaat in 24-Well-Platten zur Differenzierung von Darmorganoiden. - Spülen Sie die Zellen mit 1 mL D-PBS (ohne Ca++ und Mg++) und aspirieren Sie. 1 ml enzymfreies Reagenz für die Auswahl humaner pluripotenter Stammzellen hinzufügen, vorsichtig schwenken, um sich gleichmäßig zu verteilen, und innerhalb von 1 Minute aspirieren. Die Bienenvölker müssen nur einem dünnen Flüssigkeitsfilm ausgesetzt werden.
- Inkubieren Sie bei 37 °C, bis die Bienenvölker weniger kompakt aussehen, was ~ 3-8 Minuten dauern wird.
HINWEIS: Die optimale Inkubationszeit kann je nach verwendeter Zelllinie variieren. Die optimale Inkubationszeit muss bestimmt werden, wenn jede Zelllinie zum ersten Mal mit dem enzymfreien Reagenz zur Selektion humaner pluripotenter Stammzellen durchgelassen wird. - Fügen Sie 1 ml vollständiges Stammzellmedium hinzu. Lösen Sie die Kolonien, indem Sie vorsichtig fest auf die Seite der Platte klopfen. Achte darauf, dass du die Platte mit der anderen Hand festhältst.
- Übertragen Sie mit einer Pipette von 1 ml oder größer die Suspension der Zellklumpen in ein konisches 15-ml-Röhrchen. Bestimmen Sie die Größe der Klumpen mit einem Hellfeldmikroskop und stellen Sie sicher, dass die Größe des Zellklumpens zwischen 50 und 200 μm liegt. Wenn sie größer sind, schütteln Sie das konische Rohr vorsichtig, um sie aufzubrechen.
- Zeichnen Sie ein x-y-Gitter am Boden jeder Vertiefung einer 96-Well-Platte mit flachem Boden, die für die Klumpenzählung verwendet wird, und fügen Sie 50 μl D-PBS zu jeder Vertiefung hinzu. Es wird empfohlen, die gezählten Klumpen aus 3 Vertiefungen zu mitteln.
- Stellen Sie sicher, dass die Zellklumpen gleichmäßig verteilt sind, indem Sie das Röhrchen vorsichtig schütteln und dann 5 μl Klumpensuspension in jede der Vertiefungen der 96-Well-Platte mit flachem Boden geben.
HINWEIS: Es wird nicht empfohlen, einen automatischen Zellzähler zum Zählen von Klumpen zu verwenden. Eine manuelle Zählung wird empfohlen. - Zählen Sie die Gesamtzahl der Klumpen in jeder Vertiefung mit einem Durchmesser von 50 bis 200 μm. Wenn die meisten Zellklumpen einen Durchmesser von > 200 μm haben (siehe Abbildung 1), wiederholen Sie die Schritte 1.4.5 - 1.4.7.
- Berechnen Sie das Volumen (in μl) der Klumpensuspension, das für die Aussaat von 6.000 Klumpen erforderlich ist, wie folgt:

HINWEIS: Die optimale Aussaatdichte muss mit jeder Zelllinie optimiert werden. Um die optimale Aussaatdichte zu bestimmen, wird empfohlen, eine Erstaussaat in einer Reihe von Klumpendichten durchzuführen (z. B. 4.000, 5.000 und 6.000 Klumpen pro Well) und 24-48 Stunden nach der Aussaat zu überprüfen, ob eine Konfluenz von 85 % bis 90 % erreicht ist. - Trennen Sie zur Qualitätskontrolle eine Zellprobe für die durchflusszytometrische Untersuchung von Stammzellmarkern. In Tabelle 4 finden Sie eine Liste der gebräuchlichen Marker.
- Trennen Sie das erforderliche Volumen der iPSC-Klumpensuspension zum Saatgut auf den Wells, die mit unterschiedlichem ECM beschichtet sind, in separate 15-ml-Röhrchen.
- Das Röhrchen mit der iPSC-Klumpenlösung bei 200 x g für 5 min zentrifugieren, um Medien zu entfernen, die ein enzymfreies Reagenz zur Selektion humaner pluripotenter Stammzellen enthalten. Während Sie auf die Zentrifugation warten, fügen Sie 50 % des gewünschten Volumens pro Vertiefung zu jeder Vertiefung hinzu, falls dies noch nicht geschehen ist.
- Bereiten Sie vor dem Start folgendes vor: Mindestens 1 h vor dem Passieren die gewünschten Kunststoffartikel mit Matrizen bestreichen. Aliquotieren Sie ausreichend vollständige Stammzellmedien und erwärmen Sie sie auf Raumtemperatur (15 - 25 °C). Vermeiden Sie mehrfache Heizzyklen für das gesamte Medium.
- iPSC-Kultur in Platten, die Matrizen tierischen Ursprungs enthalten
- Resuspendieren Sie die iPS-Klumpen vorsichtig in den 50 % des berechneten Volumens des vollständigen Stammzellmediums, um die gewünschte Dichte zu erhalten, und plattieren Sie die Zellaggregatmischung auf beschichtete Wells, die 50 % des berechneten vollständigen Stammzellmediums enthalten.
HINWEIS: Die Zugabe eines ROCK-1-Inhibitors kann bei einigen Zelllinien oder bei einer Größe von 50 μm oder kleiner erforderlich sein. Es wird empfohlen, <10 μm hinzuzufügen, um eine frühe Gastrulation zu vermeiden. - Geben Sie vorsichtig Medium mit Zellen auf die Matrizen, 250 μl/Well, wenn Sie eine 24-Well-Platte verwenden. Das empfohlene Volumen des Zellmediums für Well-Platten anderer Größe finden Sie in Tabelle 2.
- Kippen Sie die Platte in mehreren kurzen Hin- und Her- und Seitwärtsbewegungen, um die Zellklumpen gleichmäßig zu verteilen.
VORSICHT: Eine ungleichmäßige Verteilung der Aggregate führt zu einer erhöhten spontanen Differenzierung der humanen iPSCs. Die kreisförmige Ausbreitung der Suspension der Klumpen führt zu Klumpen, die sich am Rand der Vertiefungen agglomerieren und in der Mitte eine geringere Dichte aufweisen. - Inkubieren Sie die Platte bei 37 °C und führen Sie täglich oder jeden zweiten Tag einen Mediumwechsel mit vollständigen Stammzellmedien durch. Wenn Sie Medienwechsel durchführen, beurteilen Sie die Kulturen visuell, um das Wachstum zu überwachen und festzustellen, ob die Zellen eine Übergabezeit benötigen oder für die Differenzierung bereit sind. Um zwei aufeinanderfolgende Fütterungstage zu überspringen, fügen Sie das 2-fache Volumen des Mediums hinzu, das für einen einzigen Tag benötigt wird.
- Resuspendieren Sie die iPS-Klumpen vorsichtig in den 50 % des berechneten Volumens des vollständigen Stammzellmediums, um die gewünschte Dichte zu erhalten, und plattieren Sie die Zellaggregatmischung auf beschichtete Wells, die 50 % des berechneten vollständigen Stammzellmediums enthalten.
- iPSC-Kultur in Platten, die Matrix 4-XE enthalten
- Resuspendieren Sie die Verklumpungen von iPSCs vorsichtig in das berechnete Gesamtvolumen von 3x vollständigen Stammzellmedien, um die gewünschte Dichte zu erhalten.
- Geben Sie vorsichtig Medium mit Zellen auf das Hydrogel 250 μl/Well, wenn Sie eine 24-Well-Platte verwenden. Das empfohlene Volumen des Zellmediums für Well-Platten anderer Größe finden Sie in Tabelle 3.
ACHTUNG: Das Hydrogel quillt auf und nimmt ein größeres Volumen ein als frisch hergestellt. iPSC-Kolonien werden teilweise in das Hydrogel am unteren Rand der Platte eingebettet, daher wird empfohlen, 50%-80% des oberen Mediums zu wechseln, ohne das Hydrogel zu stören. - Bewegen Sie die Platte in mehreren schnellen, kurzen, hin und her und von Seite zu Seite, um die Zellklumpen gleichmäßig zu verteilen.
- Stellen Sie die Platte in einen 37 °C heißen Inkubator mit 5 % CO2 und 95 % Luftfeuchtigkeit. Wechseln Sie das Zellmedium mit vollständigen Stammzellmedien. Führen Sie täglich oder jeden zweiten Tag einen Mediumwechsel durch; Für letzteres fügen Sie das 2-fache Volumen des Mediums hinzu.
- Untersuchen Sie die Kulturen visuell, um ihr Wachstum zu verfolgen, wenn sie das für die Differenzierung geeignete Stadium erreichen.

Abbildung 1: Optimale Klumpengröße. Bilder von Klumpen der iPSC-Zelllinie SCTi003A, die ein Beispiel für die optimale Klumpengröße darstellen. Maßstabsleiste = 200 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
2. hiPSC-Differenzierung und Bildung von Darmorganoiden
ACHTUNG: Alle Arbeiten werden in einer Biosicherheitswerkbank (BSC) nach aseptischen Standardtechniken durchgeführt. Muss die OSHA-Sicherheitsstandards für Labore einhalten, einschließlich der ordnungsgemäßen Verwendung von persönlicher Schutzausrüstung wie Laborkitteln, Handschuhen und Schutzbrillen.
- Bevor Sie beginnen, bereiten Sie Folgendes vor: Bereiten Sie Aliquots des Differenzierungsmediums vor, das für jede Stufe erforderlich ist. Nach dem Auftauen der Aliquote diese sofort verwenden. Nicht wieder einfrieren. Befolgen Sie Tabelle 5 für eine allgemeine Anleitung für jeden Stufenmedientyp und die Volumina, die für eine 24-Well-Platte benötigt werden.
HINWEIS: Die optimale Zeit in jeder Differenzierungsphase kann je nach Zelllinie variieren. Es wird empfohlen, die Expression jedes Zellmarkers im Stadium zu überprüfen, um die Wirksamkeit der Differenzierung zu verbessern. Die periodische Bewertung der Genexpression in den iPSCS-Kulturen war in dieser Forschung nicht enthalten, da wir herstellerqualifizierte Chargen bei geringen Passagen verwendet haben, wird jedoch als Qualitätskontrollschritt für neue/noch nicht qualifizierte iPSC-Linien während der nachfolgenden Differenzierungsschritte empfohlen. - Stufe 1: Definitives Endoderm (DE)
- Um die Bereitschaft zum Beginn der Differenzierung zu beurteilen, überprüfen Sie die folgenden Kriterien.
- Beurteilen Sie mit einem Mikroskop die Konfluenz der Stammzellkolonien und das Ausmaß der spontanen Differenzierung in der Kultur. Verlassen Sie sich nicht nur auf die morphologische Bewertung. Die optimale Konfluenz sollte zwischen 85 % und 90 % liegen (über ~105 Zellen/cm2), und die minimale spontane Differenzierung sollte <5 % betragen.
- Sammeln Sie Zellen aus einer Opfervertiefung für die durchflusszytometrische Charakterisierung von hiPSC-Markern, bevor Sie mit dem nächsten Schritt fortfahren. Für die repräsentative charakterisierte Durchflusszytometrie wurde eine Vorwärts- und Seitenstreu-Gating-Strategie verwendet. Wenn Zellen die optimalen Kriterien erfüllen, beginnen Sie mit der Differenzierung. Das Kriterium für die Zellübergabe ist >85% Expression von idealerweise 3 Markern (Tabelle 4), insbesondere bei neuen/noch nicht qualifizierten iPSC-Linien im Labor.
HINWEIS: iPSC-Kulturen, die auf unterschiedlichen Matrices gezüchtet werden, durchlaufen den gleichen Gesamtprozess, wobei nur iPSC-Wachstum auf Matrix 4-XE erfolgt, was einen schonenderen Medienwechsel erfordert, um die semi-eingebetteten Zellen nicht zu stören. Während der Induktion des definitiven Endoderms ist mit einem signifikanten Zelltod zu rechnen, da die Zellen in diesem Stadium besonders empfindlich sind. Seien Sie beim Wechsel des Mediums vorsichtig und versuchen Sie, die Zeit, die die Zellen außerhalb der 37 °C-Inkubationsumgebung ausgesetzt sind, so gering wie möglich zu halten.
- Beginnen Sie mit der Differenzierung der Zellkolonien wie unten beschrieben.
- Tag 0: Ein Aliquot von DE medium auf 37 °C erwärmen. Um eine Denaturierung von Medien durch Erwärmungs-Kühl-Zyklen zu vermeiden, stellen Sie sicher, dass nur das für Tag 0 erforderliche Volumen (0,7 ml/Well) erwärmt wird.
- Aspirieren Sie die Medien aus hiPSCs. Geben Sie vorsichtig 0,7 mL DE Medium pro Vertiefung an der Seite der Vertiefung hinunter. Vermeiden Sie hartes Pipettieren, das die Bienenvölker ablösen oder beschädigen könnte. Die Platte bei 37 °C mit 5 % CO2 und 95 % Luftfeuchtigkeit 24 h inkubieren.
- Tag 1: Erwärmen Sie ein Aliquot des DE-Mediums, das nur das für Tag 1 benötigte Volumen (0,5 mL/Well) enthält, auf 37 °C. Aspirieren Sie das DE-Medium aus den Zellen und fügen Sie vorsichtig 0,5 ml DE-Medium pro Vertiefung an der Seite der Vertiefung hinzu. Bei 37 °C mit 5 % CO2 und 95 % Luftfeuchtigkeit 24 h inkubieren.
- Tag 3: Die Zellen sind bereit, um auf die endgültige Endodermbildung untersucht zu werden. In Tabelle 4 finden Sie empfohlene Marker. Bevor Sie mit der nächsten Phase fortfahren, opfern Sie eine Vertiefung, um eine durchflusszytometrische Charakterisierung spezifischer Marker durchzuführen, die mit DE assoziiert sind. Für die repräsentative charakterisierte Durchflusszytometrie wurde eine Vorwärts- und Seitenstreu-Gating-Strategie verwendet. Es wird empfohlen, die Expression von mehr als 1 Marker zu überprüfen, insbesondere bei neuen/noch nicht qualifizierten iPSC-Linien im Labor.
- Um die Bereitschaft zum Beginn der Differenzierung zu beurteilen, überprüfen Sie die folgenden Kriterien.
- Stadium 2: Mitteldarm/Hinterdarm (MH)
- Tag 3: Erwärmen Sie nur das für Tag 3 benötigte MH-Medium-Volumen (0,5 mL/Well) auf 15 - 25 °C. DE-Medium aus den Zellen aspirieren und durch 0,5 ml MH-Medium ersetzen. Bei 37 °C mit 5 % CO2 und 95 % Luftfeuchtigkeit 24 h inkubieren.
- Tag 4 - 9: Vor dem täglichen Wechsel des Mediums verwenden Sie das Mikroskop mit 2- oder 4-facher Vergrößerung, um die Bildung von Sphäroiden zu beurteilen, indem Sie nach sichtbaren 3D-Strukturen suchen (kann ab Tag 4 passieren); frei schwebende Sphäroide (kann ab Tag 5 auftreten).
- Sobald sich die Sphäroide abzulösen beginnen, verwenden Sie eine 1-ml-Pipette, um das Medium vorsichtig von den Zellen in ein steriles konisches 15-ml-Röhrchen zu übertragen, um die Anzahl der von den Zellen abgelösten Sphäroide zu bestimmen.
HINWEIS: Vermeiden Sie Scherkräfte, die 3D-Strukturen von der Monoschicht lösen könnten. Sobald sie bereit sind, lösen sich die Sphäroide von selbst ab. - Wenn weniger als 50 Sphäroide abgelöst sind, zentrifugieren Sie bei 200 x g für 5 min, entfernen Sie das alte Medium und resuspendieren Sie die Sphäroide auf 0,5 mL/Vertiefung des Mediums und überführen Sie sie in die entsprechende Vertiefung, bis genügend Sphäroide gereift sind.
HINWEIS: Die Dauer der Exposition gegenüber dem Mitteldarm-/Hinterdarmmedium spielt eine entscheidende Rolle bei der Bestimmung der regionalen Identität der Entwicklung von Dünndarm-Organoiden, wie z. B. Duodenum (kürzere Exposition) oder Ileum (längere Exposition). Um einheitlichere Kulturen mit gleicher Identität zu erhalten, sollten Sphäroide gleichzeitig gesammelt werden, die dem Medium des Mitteldarms und des Hinterdarms ausgesetzt waren. - Wiederholen Sie Schritt 2.3.3, bis genügend Sphäroide abgelöst sind, um die Kultur des menschlichen Darmorganoids (hIO) einzubetten und zu initiieren.
- Bevor Sie mit der nächsten Phase fortfahren, trennen Sie eine Probe von Sphäroiden und führen Sie eine durchflusszytometrische Charakterisierung spezifischer Marker durch, die mit dem Mitteldarm/Hinterdarm (MH) assoziiert sind. Für die repräsentative charakterisierte Durchflusszytometrie wurde eine Vorwärts- und Seitenstreu-Gating-Strategie verwendet. In Tabelle 4 finden Sie eine Liste der häufig verwendeten Marker. Es wird empfohlen, die Expression von mehr als 1 Marker zu überprüfen, insbesondere bei neuen/noch nicht qualifizierten iPSC-Linien im Labor.
- Stufe 3: Einbettung von Sphäroiden
- Bereiten Sie Folgendes vor, bevor Sie mit dem Verfahren beginnen.
- Tauen Sie die Aliquots von Matrix 1-ABO und Matrix 3-ABO auf Eis auf. Berücksichtigen Sie die Anzahl der gesammelten Sphäroide, um die Gesamtmenge der benötigten Matrix zu bestimmen, wenn 30-40 μl Matrix pro Dom benötigt werden.
- Bereiten Sie 25 mL kaltes Advance DMEM/F-12 mit 15 mM HEPES vor. Hochkonzentrierte ECMs sind viskos und schwer zu pipettieren. Stellen Sie eine Schachtel mit sterilen 100-μl-Pipettenspitzen bei -20 °C auf, um kalte Spitzen vorzubereiten, die den Prozess unterstützen können. Bereiten Sie Aliquots von Intestinal Organoid Growth Medium (OGM) vor, für das 4 Fütterungen (0,5 ml/Well pro Fütterung) erforderlich sind. Zur Vorbereitung siehe Tabelle 5 .
- Tauen Sie ein Aliquot des OGM-Präparats auf Eis auf. Nach dem Auftauen der Aliquote diese sofort verwenden. Nicht wieder einfrieren. Lagern Sie den kompletten OGM bis zu 2 Wochen bei 2-8 °C.
- Bereiten Sie OGM vor, das 3x des Nahrungsergänzungsmittels im Vergleich zur Standardformulierung für (3x OGM) enthält. Nehmen Sie die Matrix 4-XFO1 thought Matrix 4-XFO4 aus dem Kühlschrank und lassen Sie sie auf Raumtemperatur (25 °C) erwärmen.
- Führen Sie für das System tierischen Ursprungs die unten beschriebenen Schritte aus.
- Legen Sie eine sterile 24-Well-Gewebekulturplatte in den Inkubator, um sie auf 37 °C zu erwärmen, während Sie die Sphäroide und Matrizen vorbereiten. Lassen Sie die gesammelten Sphäroide auf dem Boden eines konischen 15-ml-Röhrchens absetzen. Saugen Sie den Überstand vorsichtig an und entsorgen Sie ihn.
HINWEIS: Um einen genaueren Vergleich der Wirkung der Matrizen zu ermöglichen, stellen Sie sicher, dass die aus dem hiPSC gesammelten Sphäroide auf jeder Matrix in separaten Röhrchen differenziert aufbewahrt werden. - Geben Sie 1 mL DMEM/F-12 mit 15 mM HEPES zu den Sphäroiden. Zentrifugieren Sie bei 300 x g für 5 min bei Raumtemperatur (15 - 25 °C).
- Entfernen Sie vorsichtig so viel Überstand wie möglich. Es wird empfohlen, mit einer 1-ml-Pipettiermaschine zu beginnen und auf 100-μl- und sogar 10-μl-Pipetten umzusteigen, um so viel wie möglich zu entfernen, ohne das Pellet der Sphäroide zu stören.
HINWEIS: Je größer das verbleibende Medienvolumen ist, desto verdünnter werden die Matrizen. Verdünnte Matrizen können zu Gelierungsproblemen oder weicheren Kuppeln führen. Weichere Kuppeln führen zu einer höheren Wahrscheinlichkeit des Einsturzes bei minimalen Störungen. - Geben Sie mit einer Pipette mit einer kalten Pipettenspitze von 100 μl/50 μl/50 Sphäroide kalte (2 - 8 °C) Matrix 1-ABO oder Matrix 3-ABO in das jeweilige Röhrchen. Verteilen Sie die Sphäroide vorsichtig in der Matrix, indem Sie ~5x auf und ab pipettieren. Entleeren Sie die Pipettenspitze nicht vollständig, da dies zu Blasenbildung führen kann.
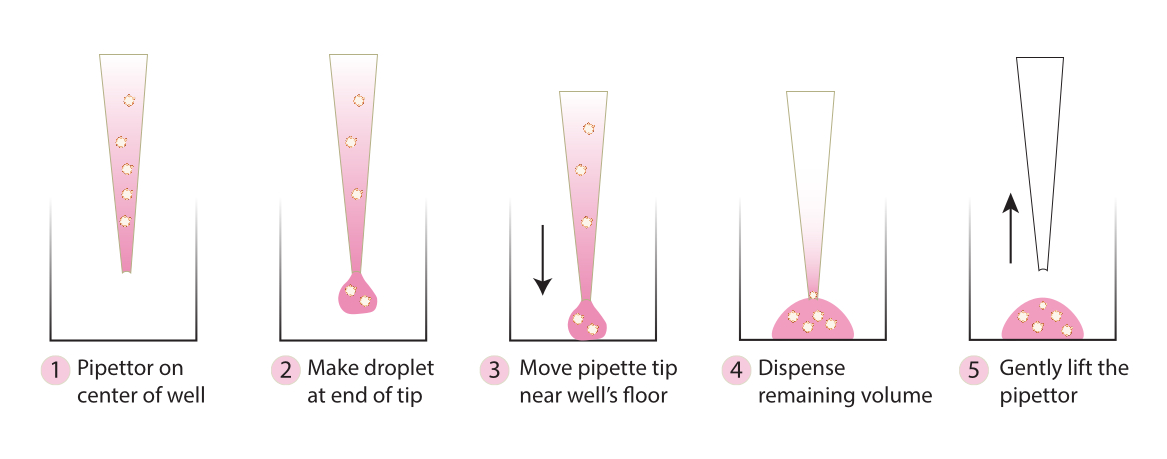
- Nehmen Sie die Platte aus dem Inkubator und übertragen Sie die eingebetteten Sphäroide mit einer kalten Pipettenspitze vorsichtig in die Mitte der Platte, indem Sie die Schritte in Abbildung 2 befolgen. Vermeiden Sie es, Matrix 1-ABO und Matrix 3-ABO zu schnell in die Kulturschale zu geben, da sonst die Kuppel abgeflacht wird.
- Schieben Sie die Platte vorsichtig in einen 37 °C heißen Inkubator und inkubieren Sie sie 30 Minuten lang, um eine Domgelierung zu gewährleisten. Seien Sie besonders vorsichtig bei Erschütterungen oder starken Bewegungen während des Transports und im Inkubator.
- Während der Inkubation der Kuppel zum Gelieren wird ein ausreichendes Volumen an intestinalem OGM (0,5 ml/Well) für die Anzahl der zu verwendenden Wells bei 37 °C erwärmt.
HINWEIS: Erwärmen Sie nur das benötigte Volumen. Vermeiden Sie Erwärmungs-Kühl-Zyklen. Stellen Sie sicher, dass das Medium warm ist, bevor Sie es in die Kuppel einlegen. Kalte Medien können zum Einsturz der Kuppel führen. - Geben Sie nach 30 Minuten vorsichtig 0,5 ml Darm-OGM an die Seite der Vertiefung, um die Kuppel nicht zu stören. Inkubieren bei 37 °C mit 5 % CO2 und 95 % Luftfeuchtigkeit. Führen Sie alle 3 - 4 Tage einen vollständigen Wechsel des Mediums durch, indem Sie das Medium entfernen und dann frisches Medium hinzufügen.
- Legen Sie eine sterile 24-Well-Gewebekulturplatte in den Inkubator, um sie auf 37 °C zu erwärmen, während Sie die Sphäroide und Matrizen vorbereiten. Lassen Sie die gesammelten Sphäroide auf dem Boden eines konischen 15-ml-Röhrchens absetzen. Saugen Sie den Überstand vorsichtig an und entsorgen Sie ihn.
- Für die Matrix 4-XF Organoid-Systeme befolgen Sie die beschriebenen Schritte.
HINWEIS: Das Matrix 4-XF-Organoidsystem umfasst vier verschiedene Optionen (O1-O4); Die Formulierung jedes Typs variiert in Bezug auf biofunktionale Liganden, Steifigkeit und Abbaubarkeit. Daher empfiehlt es sich, erste Versuche durchzuführen, um den optimalen Typ für die jeweilige Anwendung zu ermitteln. Dieses Protokoll beschreibt die Verwendung der vier Optionen, um eine optimale Formulierung für hiPSC-abgeleitete Darmorganoide zu finden. Darüber hinaus gibt es zwar mehrere Kulturprotokolle, die verwendet werden können (z. B. 3D-Verkapselung, Kuppelverkapselung), aber dieses Protokoll beinhaltet die Verwendung der Kuppelmethode, so dass es möglich ist, direkt mit den üblichen Methoden zu vergleichen, die bei der Arbeit mit tierischen Basalmembransystemen verwendet werden.- Fügen Sie 1 ml/Vertiefung Kaltvorschub DMEM/F-12 mit 16 mM HEPES zu den auf dem Matrix 4-XFO4-System erzeugten Sphäroiden hinzu. Zentrifugieren Sie bei 300 x g für 5 min bei Raumtemperatur (15 - 25 °C).
HINWEIS: Um die optimale Formulierung für die Anwendung zu finden, stellen Sie sicher, dass die Sphäroide vor der Zentrifugation auf 4 separate Röhrchen verteilt werden. Auch Sphäroide, die auf tierischen Matrizen erzeugt werden, können mit diesem System zu Darmorganoiden gereift werden. - Entfernen Sie mit einer 1-ml-Pipette den Überstand, ohne die Sphäroide zu stören. Geben Sie 50 μL 3x OGM in das Sphäroid-Pellet (~100 Sphäroide). Geben Sie 100 μl der ausgewählten Matrix 4-XFO in die 50 μl Sphäroidsuspension und mischen Sie vorsichtig 5x-10x mal. Halten Sie die Matrix 4-XFO bis 3x OGM bei einem Mischungsverhältnis von 2:1 v/v, um eine Endkonzentration von 1x zu erreichen.
- Geben Sie 40 μl des Hydrogel-Sphäroid-Gemisches in die Mitte einer 24-Well-Gewebekulturplatte. Die Platten vorsichtig in einen 37 °C heißen Inkubator geben und 30 min inkubieren.
HINWEIS: Matrix 4-XFO erfordert keine Inkubation bei 37 °C für die Gelierung; Es wird jedoch empfohlen, Organoide im Vergleich zu Matrix-1-ABO- und Matrix-3-ABO-Systemen, die für eine bessere Gelierung der Dome 37 °C benötigen, ähnlichen Bedingungen auszusetzen. - Geben Sie nach 30 Minuten vorsichtig 0,5 ml Darm-OGM an die Seite der Vertiefung, um die Kuppel nicht zu stören. Inkubieren bei 37 °C mit 5 % CO2 und 95 % Luftfeuchtigkeit. Führen Sie alle 3 - 4 Tage einen Wechsel des vollen Mediums durch, indem Sie das Medium entfernen und dann frisches Medium hinzufügen.
- Fügen Sie 1 ml/Vertiefung Kaltvorschub DMEM/F-12 mit 16 mM HEPES zu den auf dem Matrix 4-XFO4-System erzeugten Sphäroiden hinzu. Zentrifugieren Sie bei 300 x g für 5 min bei Raumtemperatur (15 - 25 °C).
- Bereiten Sie Folgendes vor, bevor Sie mit dem Verfahren beginnen.
- hIO-Passage und -Reifung
- Bereiten Sie die gleichen Chemikalien, Lösungen und Reagenzien vor wie in Schritt 2.4.1.
- Geben Sie 1-2 ml Antihaft-Spüllösung in ein konisches 15-ml-Röhrchen (1 pro Bedingung) und schwenken Sie, um das Röhrchen zu beschichten.
- Entfernen Sie die Anti-Adhärenz-Lösung und spülen Sie die Röhrchen mit 5 mL D-PBS (ohne Ca++ und Mg++). Verschließen Sie alle beschichteten Tuben und bewahren Sie sie bis zur Verwendung bei Raumtemperatur (15 - 25 °C) auf.
- Saugen Sie das Medium aus den Kuppeln an. Geben Sie mit einer 1-ml-Pipette 1 ml kaltes DMEM/F-12 direkt in die Kuppel. Ziel ist es, die Kuppeln von der Platte zu lösen.
- Geben Sie zusätzlich 1 ml kaltes DMEM/F-12 in die Vertiefung und pipettieren Sie auf und ab, um alle verbleibenden Organoide zu gewinnen. Auf die beschichteten konischen 15-ml-Röhrchen übertragen.
HINWEIS: Überprüfen Sie die erfolgreiche Ernte von Organoiden, indem Sie die Vertiefung unter einem Mikroskop visuell untersuchen. Werden Restorganoide festgestellt, ist Schritt 2.5.5 zu wiederholen. - Führen Sie mit einer 1-ml-Pipettore ein Auf- und Ab-Pipettieren der Suspension durch, um Organoide aufzulösen, bis eine gleichmäßige Fragmentsuspension mit der gewünschten Organoidgröße (z. B. 100 - 500 μm) erreicht ist.
HINWEIS: Verwenden Sie eine 200-μl-Pipette, um zu überprüfen, ob die Organoide der empfohlenen Größe entsprechen. Der Einsatz des 200-μl-Pipettierers ermöglicht bei Bedarf eine zusätzliche Fragmentierung, wodurch sichergestellt wird, dass die Fragmente reibungslos durch eine 200-μl-Pipettenspitze geleitet werden können.
ACHTUNG: Vermeiden Sie es, Fragmente durch hartes oder längeres Pipettieren in einzelne Zellen aufzubrechen. - Ermitteln Sie die gewünschte Organoiddichte, indem Sie entweder die Fragmente zählen oder das Split-Verhältnis verwenden. Trennen Sie ein Aliquot und führen Sie die Zählung mit dem gleichen Verfahren wie in den Schritten 1.4.6-1.4.9 für die Klumpenzählung durch.
HINWEIS: Die optimale Organoiddichte sollte pro Linie optimiert werden. Generell wird eine Dichte von 40 - 80 Darmorganoiden pro Dom empfohlen. - Stellen Sie sicher, dass das Röhrchen während des Organoid-Zählvorgangs auf Eis gelegt wird. Nach ca. 5 min haben sich Organoid-Fragmente aufgrund der Schwerkraft am Boden des Röhrchens abgesetzt.
HINWEIS: Je größer das Volumen der Organoidlösung ist, desto länger dauert es, bis sie sich am Boden absetzt. - Entfernen Sie vorsichtig so viel Überstand und die trübe Schicht, die sich auf den Organoiden gebildet hat. In den frühen Passagen, wenn Organoide heranreifen, umfasst diese trübe Phase eine Matrix und einzelne Zellen.
- Fügen Sie 2 mL kaltes DMEM/F-12 hinzu, indem Sie es direkt auf das Pellet pipettieren. Bei 200 x g für 5 min bei Raumtemperatur (15 - 25 °C) zentrifugieren.
- Bei Systemen tierischen Ursprungs ist der Überstand vorsichtig zu entfernen und zu entsorgen, indem die gleichen Schritte wie in den Schritten 2.4.2.4 bis 2.4.2.9 beschrieben zu befolgen sind.
- Entfernen und entsorgen Sie den Überstand für das Matrix 4-XFO-System vorsichtig gemäß den gleichen Schritten, die in den Schritten 2.4.3.2 bis 2.4.3.4 beschrieben sind.
HINWEIS: Bei der Arbeit mit dem Matrix 4-XFO-System wird empfohlen, eine xenofreie Hydrogel-Organoid-Rückgewinnungslösung für eine optimale Entfernung von Hydrogelrückständen zu verwenden. Diese Rückgewinnungslösung wird besonders bei der Umstellung von tierischen auf xenofreie Hydrogelsysteme empfohlen, um die Eliminierung von xenogenem Material zu gewährleisten.

Abbildung 2. Schematische Darstellung der für die Kuppelbildung empfohlenen Technik. Das Schema beschreibt den Schritt-für-Schritt-Prozess, der für eine erfolgreiche Kuppelbildung für alle Systeme empfohlen wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
3. Charakterisierung der E/A-Größe
HINWEIS: Die Größe der Organoide wurde durch Hellfeldbilder charakterisiert, die mit 4x und 10x aufgenommen wurden. Die Bildverarbeitungsanalyse wurde mit Hilfe von MATLAB automatisiert. Die allgemeinen Schritte des Prozesses werden im Folgenden beschrieben, und ein Beispiel des Codes ist in der ergänzenden Datei 1 enthalten.
- Definieren Sie das Verzeichnis, das die Bilder enthält, und listen Sie alle Bilddateien in diesem Verzeichnis auf. Initialisieren Sie die Tabelle, um die Ergebnisse zu speichern. Lesen Sie die Hellfeldbilder in einer Datei aus.
- Definieren Sie die Umwandlung von Pixel in μm und stellen Sie die Skala ein. Wenn das Bild keine Maßstabsleiste enthält, fordern Sie den Benutzer nach dem Umrechnungsfaktor von Pixel in μm an.
- Konvertieren Sie das Bild in Graustufen. Wenden Sie einen Gaußschen Filter an, um das Rauschen im Bild zu reduzieren. Legen Sie einen Schwellenwert für das gefilterte Bild fest, um Organoide vom Hintergrund zu trennen.
- Füllen Sie kleine Löcher und entfernen Sie kleine Objekte, um das Binärbild zu bereinigen. Führen Sie eine Analyse verbundener Komponenten durch, um einzelne Organoide zu identifizieren und ihre Eigenschaften wie Fläche, Schwerpunkt, Länge der Haupt- und Nebenachse zu berechnen.
- Berechnen Sie die Organoidgröße basierend auf den Längen der Haupt- und Nebenachse. Zeigen Sie die segmentierten Organoide auf dem Originalbild an und beschriften Sie sie mit ihrer Größe. Drucken Sie die Organoidgrößen aus und speichern Sie die Ergebnisse in einer .cvs-Datei.
HINWEIS: Die ersten Bilder wurden manuell analysiert, um die optimalen Parameter für die Standardabweichung des Gauß-Filters und den Bereich bis zum Schwellenwert zu bestimmen. Der Code in der Zusatzdatei 1 stellt ein Beispiel für das Grundgerüst für die Organoidgrößenanalyse aus Hellfeldbildern dar. Es ist jedoch eine weitere Verfeinerung erforderlich, um den spezifischen Anforderungen für jeden Bildtyp und jede Bildqualität gerecht zu werden. Der gleiche Prozess kann mit Open-Source-Software wie FIJI von Image J durchgeführt werden. - Führen Sie die folgenden Schritte aus, um die FIJI-Software zu verwenden.
- Definieren Sie die Umwandlung von Pixel in μm und stellen Sie die Skala ein. Wenn das Bild keine Maßstabsleiste enthält, fordern Sie den Benutzer nach dem Umrechnungsfaktor von Pixel in μm an, indem Sie auf Analysieren > Maßstab festlegen klicken.
- Konvertieren Sie das Bild in Graustufen, indem Sie auf Bild > Geben Sie > 8bit ein. Wenden Sie einen Gaußschen Filter an, um das Rauschen im Bild zu reduzieren, indem Sie > Gaußschen Blurb auf > Filter verarbeiten > Verwendetes Sigma (Radius): 2 klicken.
- Legen Sie einen Schwellenwert für das gefilterte Bild fest, um Organoide vom Hintergrund zu trennen, indem Sie auf Bild > Schwellenwert > MaxEntropy > Anwenden klicken.
- Füllen Sie kleine Löcher und entfernen Sie kleine Objekte, um das Binärbild zu bereinigen, indem Sie auf > Rauschen verarbeiten > Ausreißer entfernen > 20 Pixel klicken.
- Führen Sie eine Analyse verbundener Komponenten durch, um einzelne Organoide zu identifizieren und ihre Eigenschaften wie Fläche, Schwerpunkt, Länge der Hauptachse und Länge der Nebenachse zu berechnen, indem Sie auf Analysieren > Messwert festlegen > Klicken Sie darauf, dass Fläche, Umfang und Durchmesser > Analysieren > Partikel analysieren > Umrisse anzeigen enthalten sind.
- Statistische Analyse
- Bewerten Sie die Datenverteilung auf Normalität durch den Saphiro-Wilk-Test mit der Software JMP (SAS). Um statistische Unterschiede zwischen Gruppen zu untersuchen, führen Sie eine bidirektionale ANOVA durch und führen Sie Post-hoc-Tests mit der nichtparametrischen Wilcoxon-Methode in der JMP(SAS)-Software durch. Die Signifikanz wurde bei einem Alpha-Niveau von p ≤ 0,05 ermittelt.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Nach diesem Protokoll wurden kommerziell erhältliche Basalmembranen und ein xenofreies Hydrogelsystem erfolgreich eingesetzt, um hiPSC-Zellen zu kultivieren und sie in hIO zu differenzieren. Das Hauptziel dieser Experimente war es, die Äquivalenz von Matrizen aus verschiedenen Quellen für die hiPSC- und hIO-Arbeit systematisch zu evaluieren. Der erste Abschnitt dieses Protokolls konzentrierte sich auf die Aufrechterhaltung und Charakterisierung einer gesunden iPSC-Kultur, die zu einer effizienten Generierung von Darmo...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Die Auswahl der optimalen Mikroumgebung für die Arbeit mit Stammzellen und Organoiden ist ein entscheidender erster Schritt beim Einsatz dieser Plattformen für eine Vielzahl von Anwendungen. Unsere repräsentativen Ergebnisse zeigen, dass Matrix 4-XFO3 in Kombination mit einer höheren Konzentration an Wachstumsfaktoren zu größeren Organoiden führt, was darauf hindeutet, dass die physikalischen Eigenschaften von xenofreien Hydrogelen genutzt werden können, um die Organoidbildung mit diesen Systemen zu optimieren. E...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Dr. John Huang ist Gründer und CEO von TheWell Bioscience.
Danksagungen
Die Autoren würdigen frühere Schulungen und allgemeine Empfehlungen zum Beginn der hiPSC- und Organoid-Arbeit von Dr. Christina Pacak, Silveli Susuki-Hatano und Russell D'Souza. Sie danken Dr. Chelsey Simmons für ihre Anleitung bei der Verwendung von Hydrogelsystemen für die In-vitro-Zellkulturarbeit . Die Autoren danken auch Dr. Christine Rodriguez und Dr. Thomas Allison von STEMCELL Technologies für ihre Anleitung zur hiPS-Kultur. Die Autoren danken auch TheWell Bioscience für die Übernahme der Publikationskosten.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 24-Well Plate (Culture treated, sterile) | Falcon | 353504 | |
| 37 °C water bath | VWR | ||
| 96-well plate | Fisher Scientific | FB012931 | |
| Advanced DMEM/F12 | Life Technologies | 12634 | |
| Anti-adherence Rinsing Solutio | STEMCELL Technologies | 7010 | |
| Biological safety cabinet (BSC) | Labconco | Logic | |
| Brightfield Microscope | Echo Rebel | REB-01-E2 | |
| BXS0116 | ATCC | ACS-1030 | |
| Centrifuge with temperature control (4 °C capabilities) | ThermoScientific | 75002441 | |
| Conical tubes, 15 mL, sterile | Thermo Fisher Scientific | 339650 | |
| Conical tubes, 50 mL, sterile | Thermo Fisher Scientific | 339652 | |
| Cultrex RGF BME, Type 2 | Bio-techne | 3533-005-02 | |
| Cultrex Stem Cell Qualified RGF BME | Bio-techne | 3434-010-02 | |
| D-PBS (Without Ca++ and Mg++) | Thermo Fisher Scientific | 14190144 | |
| GeltrexLDEV-Free, hESC-Qualified Reduce Growth Factor | Gibco | A14133-02 | |
| GlutaMAX Supplement | Thermo Fischer Scientific | 35050-061 | |
| Guava Muse Cell Analyzer or another flow cytometry equipment (optional) | Luminex | 0500-3115 | |
| HEPES buffer solution | Thermo Fischer Scientific | 15630-056 | |
| Heralcell Vios Cell culture incubator (37 °C, 5% CO2) | Thermo Scientific | 51033775 | |
| JMP Software | SAS Institute | JMP 16 | |
| MATLAB | MathWorks, Inc | R2022b | |
| Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix LDEV free | Corning | 356231 | |
| Matrigel Matrix High Concentration (HC), Growth Factor Reduced (GFR) LDEV-free | Corning | 354263 | |
| mTeSR Plus Medium | STEMCELL Technologies | 100-0276 | |
| Nunclon Delta surface treated 24-well plate | Thermo Scientific | 144530 | |
| PE Mouse Anti-human CD326 (EpCAM) | BD Pharmingen | 566841 | |
| PE Mouse Anti-human CDX2 | BD Pharmingen | 563428 | |
| PE Mouse Anti-human FOXA2 | BD Pharmingen | 561589 | |
| PerCP-Cy 5.5 Mouse Anti-human SSEA4 | BD Pharmingen | 561565 | |
| ReLeSR | STEMCELL | 5872 | |
| SCTi003-A | STEMCELL Technologies | 200-0510 | |
| Serological pipettes (10 mL) | Fisher Scientific | 13-678-11E | |
| Serological pipettes (5 mL) | Fisher Scientific | 13-678-11D | |
| STEMdiff Intestinal Organoid Growth Medium | STEMCELL Technologies | 5145 | |
| STEMdiff Intestinal Organoid Kit | STEMCELL Technologies | 5140 | |
| Vitrogel Hydrogel Matrix | TheWell Bioscience | VHM01 | |
| VitroGel ORGANOID Discovery Kit | TheWell Bioscience | VHM04-K |
Referenzen
- Hynes, R. O. Integrins: Bidirectional, allosteric signaling machines. Cell. 110 (6), 673-687 (2002).
- Frantz, C., Stewart, K. M., Weaver, V. M. The extracellular matrix at a glance. J Cell Sci. 123, Pt 24 4195-4200 (2010).
- Hinz, B., Gabbiani, G. Fibrosis: Recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol Rep. 2, 78(2010).
- Pickup, M. W., Mouw, J. K., Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15 (12), 1243-1253 (2014).
- Rozario, T., DeSimone, D. W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev Biol. 341 (1), 126-140 (2010).
- Even-Ram, S., Artym, V., Yamada, K. M. Matrix control of stem cell fate. Cell. 126 (4), 645-647 (2006).
- Engler, A. J., Sen, S., Sweeney, H. L., Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell. 126 (4), 677-689 (2006).
- Tran, O. N., et al. Organ-specific extracellular matrix directs trans-differentiation of mesenchymal stem cells and formation of salivary gland-like organoids in vivo. Stem Cell Res Ther. 13 (1), 306(2022).
- Nikolaev, M., et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature. 585 (7826), 574-578 (2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., et al. Tissue geometry drives deterministic organoid patterning. Science. 375 (6576), (2022).
- Heo, J. H., Kang, D., Seo, S. J., Jin, Y. Engineering the extracellular matrix for organoid culture. Int J Stem Cells. 15 (1), 60-69 (2022).
- Shamir, E. R., Ewald, A. J. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 15 (10), 647-664 (2014).
- Clevers, H. Modeling development and disease with organoids. Cell. 165 (7), 1586-1597 (2016).
- Jung, P., et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 17 (10), 1225-1227 (2011).
- Lancaster, M. A., Knoblich, J. A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science. 345 (6194), 1247125(2014).
- Huch, M., et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 494 (7436), 247-250 (2013).
- Greenlee, A. R., Kronenwetter-Koepel, T. A., Kaiser, S. J., Liu, K. Comparison of Matrigel and gelatin substrata for feeder-free culture of undifferentiated mouse embryonic stem cells for toxicity testing. Toxicol In Vitro. 19 (3), 389-397 (2005).
- Geltrex LDEV-Free, HESC-Qualified, Reduced Growth Factor Basement Membrane Matrix User Guide (Pub.No. MAN0007336 3.0. Fisher Scientific. , Available from: https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.cn/TFS-Assets%2FLSG%2Fmanuals%2FGeltrex_LDEV_Free_hESC_qualified_PI.pdf (2024).
- biotechne R&D Systems. Cultrex Stem Cell Qualified Reduced Growth Factor. biotechne R&D Systems. , (2024).
- VitroGel Organoid Protocol. TheWell Bioscience. , Available from: https://www.thewellbio.com/video-protocols (2024).
- Spence, J. R., et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 470 (7332), 105-110 (2011).
- Henderson, J. K., et al. Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells. 20 (4), 329-337 (2002).
- Haruna, N. F., Huang, J. Investigating the dynamic biophysical properties of a tunable hydrogel for 3D cell culture. J Cytol Tissue Biol. 7, 30(2020).
- Cherne, M. D., et al. A synthetic hydrogel, VitroGel ORGANOID-3, improves immune cell-epithelial interactions in a tissue chip co-culture model of human gastric organoids and dendritic cells. Front Pharmacol. 12, 707891(2021).
- Stewart, D. C., et al. Quantitative assessment of intestinal stiffness and associations with fibrosis in human inflammatory bowel disease. PLoS One. 13, e0200377(2018).
- Hernandez-Gordillo, V., et al. Fully synthetic matrices for in vitro culture of primary human intestinal enteroids and endometrial organoids. Biomaterials. 254, 120125(2020).
- Broguiere, N., et al. Growth of epithelial organoids in a defined hydrogel. Adv Mater. 30, 1801621(2018).
- Barthes, J., et al. Cell microenvironment engineering and monitoring for tissue engineering and regenerative medicine: The recent advances. BioMed Res Int. 2014, 921905(2014).
- Engler, A. J., Sen, S., Sweeney, H. L., Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell. 126 (4), 677-689 (2006).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nat Rev Mater. 5 (7), 539-551 (2020).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten