
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Etude comparative des matrices de membrane basale pour l’entretien des cellules souches humaines et la génération d’organoïdes intestinaux
Dans cet article
Résumé
Les organoïdes sont devenus des outils précieux pour la modélisation des maladies. La matrice extracellulaire (MEC) guide le destin cellulaire pendant la génération d’organoïdes, et l’utilisation d’un système qui ressemble au tissu natif peut améliorer la précision du modèle. Cette étude compare la génération d’organoïdes intestinaux humains dérivés de cellules souches pluripotentes induites dans des ECM d’origine animale et des hydrogels sans xéno.
Résumé
La matrice extracellulaire (MEC) joue un rôle essentiel dans le comportement et le développement cellulaires. Les organoïdes générés à partir de cellules souches pluripotentes induites humaines (hiPSC) sont sous les feux de la rampe dans de nombreux domaines de recherche. Cependant, l’absence de repères physiologiques dans les matériaux de culture cellulaire classiques entrave la différenciation efficace des iPSC. L’incorporation de la MEC disponible dans le commerce dans la culture de cellules souches fournit des indices physiques et chimiques bénéfiques pour la maintenance cellulaire. Les produits de membrane basale d’origine animale disponibles dans le commerce sont composés de protéines ECM et de facteurs de croissance qui soutiennent le maintien des cellules. Étant donné que l’ECM possède des propriétés spécifiques aux tissus qui peuvent moduler le destin cellulaire, des matrices sans xéno sont utilisées pour diffuser la traduction vers les études cliniques. Bien que les matrices disponibles dans le commerce soient largement utilisées dans les travaux sur les hiPSC et les organoïdes, l’équivalence de ces matrices n’a pas encore été évaluée. Ici, une étude comparative de l’entretien des hiPSC et de la génération d’organoïdes intestinaux humains (hIO) dans quatre matrices différentes : Matrigel (Matrix 1-AB), Geltrex (Matrix 2-AB), Cultrex (Matrix 3-AB) et VitroGel (Matrix 4-XF) a été menée. Bien que les colonies n’aient pas une forme parfaitement ronde, il y avait une différenciation spontanée minimale, avec plus de 85% des cellules exprimant le marqueur de cellules souches SSEA-4. La matrice 4-XF a conduit à la formation de touffes rondes en 3D. De plus, l’augmentation de la concentration du supplément et des facteurs de croissance dans les milieux utilisés pour fabriquer la solution d’hydrogel Matrix 4-XF a amélioré l’expression de SSEA-4 par 1,3. La différenciation de l’hiPSC maintenue par la matrice 2-AB a entraîné moins de libérations de sphéroïdes au cours du stade de l’intestin moyen/postérieur par rapport aux autres membranes basales d’origine animale. Par rapport à d’autres, la matrice d’organoïdes sans xéno (matrice 4-O3) conduit à des hIO plus grands et plus matures, ce qui suggère que les propriétés physiques des hydrogels sans xénogrammes peuvent être exploitées pour optimiser la génération d’organoïdes. Dans l’ensemble, les résultats suggèrent que les variations dans la composition des différentes matrices affectent les étapes de différenciation des IO. Cette étude sensibilise aux différences entre les matrices disponibles dans le commerce et fournit un guide pour l’optimisation des matrices lors des travaux iPSC et IO.
Introduction
La matrice extracellulaire (MEC) est un composant dynamique et multifonctionnel des tissus qui joue un rôle central dans la régulation du comportement et du développement cellulaires. En tant que réseau complexe, il fournit un support structurel, des ligands adhésifs cellulaires1 et le stockage de facteurs de croissance et de cytokines qui régulent la signalisation cellulaire. Par exemple, lors de la cicatrisation des plaies, l’ECM sert d’échafaudage pour les cellules migrantes et de réservoir de facteurs de croissance impliqués dans la réparation tissulaire2. De même, la dérégulation de l’ECM peut entraîner une augmentation de la gravité de diverses maladies telles que la fibrose et le cancer 3,4. Au cours du développement embryonnaire, la MEC guide la morphogenèse des tissus. Par exemple, dans le développement du cœur, les composants de l’ECM jouent un rôle dans la création de l’architecture et de la fonction correctes du tissu cardiaque5. Plus d’une décennie de recherche a montré que la rigidité du microenvironnement à elle seulepeut contrôler la spécification de la lignée des cellules souches. Par conséquent, il n’est pas surprenant qu’au cours de la différenciation cellulaire in vitro, l’ECM influence le destin des cellules souches en fournissant des signaux de différenciation.
Les organoïdes peuvent être générés à partir de cellules souches pluripotentes induites (iPSC). Il est nécessaire de commencer par une lignée d’iPSC correctement caractérisée pour générer des organoïdes avec succès. Cependant, l’absence de signaux physiologiques dans les matériaux de culture cellulaire classiques entrave la différenciation efficace des iPSC et la génération d’organoïdes. De plus, des recherches récentes ont souligné l’importance de la composition de la matrice extracellulaire (MEC), des interactions entre les cellules et la MEC8, ainsi que des indices mécaniques et géométriques 9,10,11 dans le contexte de l’expansion et de la différenciation des organoïdes12. Pour faire progresser la technologie des organoïdes en améliorant la reproductibilité, il faudra incorporer des indices physiques et chimiques spécifiques aux tissus.
Les organoïdes visent à récapituler le tissu natif dans un microenvironnement physiologiquement similaire. Le choix d’un système ECM qui imite étroitement l’ECM des tissus natifs est crucial pour obtenir une pertinence physiologique concernant le comportement cellulaire, la fonction et la réponse aux stimuli13. Le choix des composants de la MEC peut influencer la différenciation des cellules souches en types de cellules spécifiques au sein de l’organoïde. Différentes protéines ECM et leurs combinaisons peuvent fournir des indices qui guident le destin cellulaire14. Par exemple, des études ont montré que l’utilisation de composants ECM spécifiques peut favoriser la différenciation des cellules souches intestinales en types de cellules intestinales matures, ce qui permet d’obtenir des organoïdes intestinaux physiologiquement pertinents15. Bien que les organoïdes soient un outil précieux lors de la modélisation des maladies et des tests de médicaments, le choix d’un système ECM approprié est essentiel à cette application. Un système ECM approprié peut améliorer la précision de la modélisation de la maladie en créant un microenvironnement qui ressemble au tissu affecté16. De plus, la MEC spécifique aux tissus peut aider à générer des organoïdes qui récapitulent mieux les phénotypes associés à la maladie et les réponses aux médicaments17. L’optimisation du système ECM utilisé dans la différenciation des organoïdes est essentielle pour obtenir les résultats de différenciation souhaités.
Les systèmes de membrane basale disponibles dans le commerce dérivés de sources ECM animales (par exemple, Matrigel, Cultrex) et d’hydrogel sans xénogrammes (par exemple, VitroGel) sont largement utilisés dans la recherche sur les iPSC et les organoïdes. Les entreprises qui les commercialisent et les chercheurs qui les utilisent ont élaboré de nombreuses instructions pour leurs produits et applications spécifiques au fil des ans. Beaucoup de ces instructions ont servi de guide pour la génération de ce protocole. De plus, les avantages et les inconvénients associés à leurs propriétés intrinsèques ont été notés individuellement par de nombreux 18,19,20,21. Cependant, il n’existe pas de flux de travail systématique pour guider la sélection des systèmes optimaux pour le travail des iPSC et des organoïdes. Ici, un flux de travail permettant d’évaluer systématiquement l’équivalence des systèmes ECM provenant de diverses sources pour le travail sur les iPSC et les organoïdes est fourni. Il s’agit d’une étude comparative du maintien de la génération de deux lignées différentes d’iPSC humaines (hiPSC) et d’organoïdes intestinaux humains (hIO) dans quatre matrices différentes : Matrigel (Matrix 1-AB), Geltrex (Matrix 2-AB), Cultrex (Matrix 3-AB) et VitroGel (Matrix 4-XF). Pour la culture d’organoïdes, quatre versions de la matrice sans xéno-xéno-VitroGel qui avaient été préalablement optimisées pour la culture d’organoïdes ont été utilisées : ORGANOID 1 (matrice 4-O1), ORGANOID 2 (matrice 4-O2), ORGANOID 3 (matrice 4-O3), ORGANOID 4 (matrice 4-O4). De plus, des matrices d’origine animale optimisées pour les organoïdes ont été utilisées : Matrigel High Concentration (Matrix 1-ABO) et Cultrex Type 2 (Matrix 3-ABO). Des milieux de culture de cellules souches disponibles dans le commerce (mTeSR Plus) et un kit de différenciation des organoïdes (STEMdiff intestinal organoïdes kit) ont été utilisés. Ce protocole combine les instructions individuelles des fabricants de produits avec des expériences de laboratoire pour guider le lecteur vers une optimisation réussie de l’ECM pour leur travail spécifique sur les iPSC et les organoïdes. Dans l’ensemble, ce protocole et les résultats représentatifs soulignent l’importance de sélectionner le microenvironnement optimal pour le travail sur les cellules souches et la différenciation des organoïdes.
Protocole
1. Maintenance des hiPSC
ATTENTION : Tous les travaux sont effectués dans une enceinte de biosécurité (BSC) selon des techniques aseptiques standard. Doit suivre les normes de sécurité de l’OSHA pour les laboratoires, y compris l’utilisation appropriée d’équipements de protection individuelle tels que des blouses de laboratoire, des gants et des lunettes de protection.
- Préparation de matrices, d’aliquotes et de milieux de culture cellulaire
- Pour les membranes basales d’origine animale disponibles dans le commerce (BM ; Matrice 1-AB, Matrice 2-AB, Matrice 3-AB), préparer des aliquotes des volumes de travail en suivant les recommandations du fabricant résumées dans le tableau 1 et les stocker à -20 °C ou -80 °C pour un stockage à long terme. Évitez la formation de bulles. Si des bulles se forment, avant de les stocker, centrifuger les échantillons à 4 °C, à <200 x g, pendant ~1-2 min pour forcer la bulle à remonter à la surface.
REMARQUE : La fabrication d’aliquotes à usage unique permet d’éviter les cycles répétés de gel-dégel qui perturbent l’architecture ECM. Étant donné que la concentration de BM varie en fonction du numéro de lot, assurez-vous de suivre les recommandations du fabricant pour préparer des aliquotes à usage unique et fabriquer des solutions de revêtement. Les BM à haute concentration sont visqueux et difficiles à pipeter ; utilisez des pointes froides préalablement stockées à -20 °C. - Préparez le milieu de culture de cellules souches en suivant les recommandations du fabricant. Pour préparer le milieu complet spécifique disponible dans le commerce utilisé dans ce protocole, ajoutez 100 mL de cocktail 5x supplément à 400 mL de milieu basal. Ensuite, mélangez soigneusement ce milieu et aliquote dans des volumes de 40 ml, conservés à 20 °C. Pour l’utilisation, décongelez chaque aliquote du milieu complet, utilisez immédiatement le milieu complet aliquote ou conservez-le entre 2 et 8 °C jusqu’à 2 semaines. Ne le recongelez pas.
- Préparez des aliquotes du milieu complet de cellules souches contenant 3 fois la concentration du cocktail de suppléments pour préparer l’hydrogel Matrix 4-XE, c’est-à-dire diluez 5 fois le supplément à 3 fois.
- Pour les membranes basales d’origine animale disponibles dans le commerce (BM ; Matrice 1-AB, Matrice 2-AB, Matrice 3-AB), préparer des aliquotes des volumes de travail en suivant les recommandations du fabricant résumées dans le tableau 1 et les stocker à -20 °C ou -80 °C pour un stockage à long terme. Évitez la formation de bulles. Si des bulles se forment, avant de les stocker, centrifuger les échantillons à 4 °C, à <200 x g, pendant ~1-2 min pour forcer la bulle à remonter à la surface.
- Revêtement de plastique de culture tissulaire avec des BM d’origine animale (Matrix 1-3AB)
- Préparez ce qui suit avant de commencer : conservez toujours les matrices dans de la glace lors de la décongélation et de la manipulation pour éviter qu’elles ne gèlent. Utilisez un milieu froid pour préparer les matrices diluées. Préparez suffisamment de puits de réplication par condition pour analyser les cellules avant de commencer et à chaque étape du processus de différenciation des hiPSC et de génération d’organoïdes hIO.
REMARQUE : Ici, des plaques à 24 puits ont été utilisées pour cette étude. Reportez-vous au tableau 2 pour les volumes de revêtement recommandés pour les autres tailles de plaques. - Pour chaque type de matrice, préparer 25 mL de DMEM/F-12 à avance froide contenant 15 mM de HEPES dans un tube conique de 50 mL. Gardez-les sur de la glace.
- Ajoutez les matrices décongelées à leur avance à froid DMEM/F-12 respective et mélangez bien. Gardez le support froid pendant le processus de mélange. Vérifiez visuellement l’homogénéité du mélange en vous assurant qu’il n’y a pas de grumeaux.
- Utiliser immédiatement les solutions matricielles diluées pour enrober chaque puits choisi pour l’utilisation (250 μL/puits si vous utilisez des plaques à 24 puits).
- Inclinez doucement la vaisselle de culture pour répartir uniformément la solution d’enrobage sur toute la surface. Incuber à température ambiante (15 - 25 °C) pendant au moins 1 h avant utilisation.
REMARQUE : Si elle n’est pas utilisée immédiatement, la plaque peut être stockée à 2 - 8 °C jusqu’à 1 semaine après le revêtement, mais elle doit être scellée avec un film transparent pour éviter l’évaporation. Lorsque vous utilisez des plaques stockées, laissez-les atteindre la température ambiante (15 - 25 °C) pendant 30 minutes avant de passer à l’étape suivante. - Prélever délicatement l’excédent de solution à l’aide d’une pipette sérologique ou par aspiration. Assurez-vous que la surface revêtue n’est pas rayée.
- Ajouter immédiatement un milieu de cellules souches complet chaud (50 % du volume total nécessaire pour un puits spécifique, p. ex., 250 μL/puits si vous utilisez une plaque à 24 puits).
- Préparez ce qui suit avant de commencer : conservez toujours les matrices dans de la glace lors de la décongélation et de la manipulation pour éviter qu’elles ne gèlent. Utilisez un milieu froid pour préparer les matrices diluées. Préparez suffisamment de puits de réplication par condition pour analyser les cellules avant de commencer et à chaque étape du processus de différenciation des hiPSC et de génération d’organoïdes hIO.
- Revêtement de plastique de culture tissulaire avec l’hydrogel Matrix 4-XE
- Préparez les éléments suivants avant de commencer : sortez l’hydrogel Matrix 4-XE du réfrigérateur et laissez-le chauffer à température ambiante (25 °C). Préparez suffisamment de puits de réplication par condition pour analyser les cellules avant de commencer et à chaque étape du processus de différenciation des hiPSC et de génération d’organoïdes hIO.
REMARQUE : Ici, des plaques à 24 puits ont été utilisées pour cette étude. Reportez-vous au tableau 3 pour les volumes de revêtement recommandés pour les autres tailles de plaques. - Préparez un milieu de culture de cellules souches complet contenant une concentration de facteur de croissance 3x par rapport à la formulation standard pour la culture de cellules souches (milieu de cellules souches 3x).
- Mélangez l’hydrogel Matrix 4-XE et le milieu de cellules souches 3x à un rapport de mélange v/v de 2:1 et pipetez doucement de haut en bas 5x-10x pour bien mélanger.
- Transférez le mélange d’hydrogel dans une assiette à puits et inclinez soigneusement la vaisselle de culture pour répartir le mélange uniformément sur la surface. Utilisez 250 μL/puits si vous utilisez une plaque à 24 puits. Voir le tableau 3 pour les volumes recommandés.
- Attendre 10-15 min à température ambiante pour la formation de gel mou. Pendant le processus de formation de l’hydrogel, ne perturbez pas l’hydrogel en inclinant ou en secouant la plaque.
- Préparez les éléments suivants avant de commencer : sortez l’hydrogel Matrix 4-XE du réfrigérateur et laissez-le chauffer à température ambiante (25 °C). Préparez suffisamment de puits de réplication par condition pour analyser les cellules avant de commencer et à chaque étape du processus de différenciation des hiPSC et de génération d’organoïdes hIO.
- Passage et ensemencement des touffes sans enzymes hiPSC
- Préparez ce qui suit avant de commencer : au moins 1 h avant de passer, enduire la vaisselle en plastique souhaitée avec des matrices. Prélever suffisamment de milieu de cellules souches complet et chauffer à température ambiante (15 - 25 °C). Évitez plusieurs cycles de chauffage pour l’ensemble du support.
REMARQUE : Les étapes ci-dessous décrivent le passage d’une culture confluente déjà établie et à >90 % de CSPi sur une plaque à 6 puits et leur ensemencement dans des plaques à 24 puits pour la différenciation des organoïdes intestinaux. - Rincer les cellules avec 1 mL de D-PBS (sans Ca++ et Mg++) et aspirer. Ajouter 1 ml de réactif de sélection de cellules souches pluripotentes humaines sans enzymes, agiter soigneusement pour s’étaler uniformément et aspirer dans les 1 min. Les colonies n’ont besoin d’être exposées qu’à une fine pellicule de liquide.
- Incuber à 37 °C jusqu’à ce que les colonies commencent à paraître moins compactes, ce qui prendra ~ 3-8 min.
REMARQUE : Le temps d’incubation optimal peut varier en fonction de la lignée cellulaire utilisée. Le temps d’incubation optimal doit être déterminé lors du premier passage de chaque lignée cellulaire avec le réactif de sélection de cellules souches pluripotentes humaines exemptes d’enzymes. - Ajouter 1 mL de milieu de cellules souches complet. Détachez les colonies en tapotant soigneusement et fermement sur le côté de la plaque. Assurez-vous de tenir l’assiette avec l’autre main.
- À l’aide d’une pipette de 1 mL ou plus, transférez la suspension de cellules agglomérées dans un tube conique de 15 mL. Évaluez la taille des amas à l’aide d’un microscope à fond clair et assurez-vous que la taille de l’amas cellulaire est comprise entre 50 et 200 μm ; S’ils sont plus gros, secouez doucement le tube conique pour les décomposer.
- Dessinez une grille x-y au fond de chaque puits d’une plaque à fond plat de 96 puits qui sera utilisée pour le comptage des amas et ajoutez 50 μL de D-PBS dans chacun des puits. Il est recommandé de faire la moyenne des touffes comptées à partir de 3 puits.
- Assurez-vous que les amas cellulaires sont répartis uniformément en secouant doucement le tube, puis transférez 5 μL de suspension agglomérante dans chacun des puits de la plaque à fond plat à 96 puits.
REMARQUE : Il n’est pas recommandé d’utiliser un compteur automatique de cellules pour compter les amas Un comptage manuel est recommandé. - Comptez le nombre total d’amas de 50 à 200 m de diamètre dans chaque puits. Si la plupart des amas cellulaires ont > 200 m de diamètre (voir la figure 1), répétez les étapes 1.4.5 à 1.4.7.
- Calculez le volume (en μL) de suspension en mottes nécessaire pour ensemencer 6 000 mottes comme suit :

REMARQUE : La densité de semis optimale doit être optimisée pour chaque lignée cellulaire. Pour déterminer la densité de semis optimale, il est recommandé d’effectuer un premier semis dans une gamme de densités de touffes (p. ex., 4 000, 5 000 et 6 000 touffes par puits) et de vérifier 24 à 48 h après le semis si la confluence de 85 % à 90 % est atteinte. - Dans le cadre du contrôle de la qualité, séparer un échantillon de cellules pour l’examen par cytométrie en flux des marqueurs de cellules souches. Reportez-vous au tableau 4 pour une liste des marqueurs courants.
- Séparer le volume requis de suspension de touffes de CSPi pour semer dans les puits recouverts de différents ECM dans des tubes séparés de 15 ml.
- Centrifuger le tube contenant la solution agglomérée d’iPSC à 200 x g pendant 5 min pour prélever le milieu contenant le réactif de sélection de cellules souches pluripotentes humaines exemptes d’enzymes. En attendant la centrifugation, ajoutez 50% du volume souhaité par puits dans chaque puits si ce n’est pas déjà fait.
- Préparez ce qui suit avant de commencer : au moins 1 h avant de passer, enduire la vaisselle en plastique souhaitée avec des matrices. Prélever suffisamment de milieu de cellules souches complet et chauffer à température ambiante (15 - 25 °C). Évitez plusieurs cycles de chauffage pour l’ensemble du support.
- Culture d’iPSC dans des plaques contenant des matrices d’origine animale
- Remettez doucement en suspension les amas d’iPSC dans le volume calculé de 50 % de milieu de cellules souches complètes pour obtenir la densité souhaitée et placez le mélange d’agrégats cellulaires sur des puits enrobés contenant 50 % de milieux de cellules souches complètes calculés.
REMARQUE : L’ajout d’un inhibiteur de ROCK-1 peut être nécessaire pour certaines lignées cellulaires ou lorsque les amas mesurent 50 μm ou moins. Il est recommandé d’ajouter <10 μm pour éviter une gastrulation précoce. - Ajouter délicatement le milieu avec des cellules sur les matrices, 250 μL/puits si vous utilisez une plaque à 24 puits. Pour connaître le volume recommandé de milieu cellulaire pour les plaques à puits d’autres tailles, consultez le tableau 2.
- Inclinez la plaque en plusieurs courts mouvements d’avant en arrière et d’un côté à l’autre pour répartir uniformément les amas de cellules.
ATTENTION : La distribution inégale des agrégats entraîne une différenciation spontanée accrue des CSPi humaines. L’étalement circulaire de la suspension des touffes entraîne l’agglomération des touffes en bordure des puits et une diminution de la densité au centre. - Incuber la plaque à 37 °C et effectuer des changements de milieu à l’aide d’un milieu de cellules souches complet tous les jours ou tous les deux jours. Lorsque vous effectuez des changements de milieu, évaluez visuellement les cultures pour surveiller la croissance et déterminer si les cellules nécessitent un temps de passage ou sont prêtes pour la différenciation. Pour sauter deux jours consécutifs de tétée, ajoutez 2 fois le volume de milieu nécessaire pour une seule journée.
- Remettez doucement en suspension les amas d’iPSC dans le volume calculé de 50 % de milieu de cellules souches complètes pour obtenir la densité souhaitée et placez le mélange d’agrégats cellulaires sur des puits enrobés contenant 50 % de milieux de cellules souches complètes calculés.
- Culture d’iPSC dans des plaques contenant Matrix 4-XE
- Remettez doucement en suspension les agglomérations d’iPSC dans le volume total calculé de 3 milieux de cellules souches complets pour obtenir la densité souhaitée.
- Ajouter délicatement le milieu avec des cellules sur l’hydrogel 250 μL/puits si vous utilisez une plaque à 24 puits. Pour connaître le volume recommandé de milieu cellulaire pour les plaques à puits d’autres tailles, consultez le tableau 3.
ATTENTION : L’hydrogel gonflera et occupera un volume plus important que celui fraîchement préparé. Les colonies d’iPSC seront partiellement intégrées dans l’hydrogel au bas de la plaque, il est donc recommandé de changer 50 à 80 % du milieu supérieur sans perturber l’hydrogel. - Déplacez la plaque en plusieurs mouvements rapides, courts, d’avant en arrière et d’un côté à l’autre pour répartir uniformément les amas de cellules.
- Placez la plaque dans un incubateur à 37 °C avec 5% de CO2 et 95% d’humidité. Changez le milieu cellulaire à l’aide d’un milieu de cellules souches complet. Effectuez des changements moyens tous les jours ou tous les deux jours ; Pour ce dernier, ajoutez 2x le volume du support.
- Inspectez visuellement les cultures pour suivre leur croissance à mesure qu’elles atteignent le stade propice à la différenciation.

Figure 1 : Taille optimale des touffes. Images d’amas de lignées cellulaires d’iPSC SCTi003A illustrant un exemple de taille optimale d’amas. Barre d’échelle = 200 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Différenciation des hiPSC et génération d’organoïdes intestinaux
ATTENTION : Tous les travaux sont effectués dans une enceinte de biosécurité (BSC) selon des techniques aseptiques standard. Doit suivre les normes de sécurité de l’OSHA pour les laboratoires, y compris l’utilisation appropriée d’équipements de protection individuelle tels que des blouses de laboratoire, des gants et des lunettes de protection.
- Préparez les éléments suivants avant de commencer : préparez les aliquotes du milieu de différenciation requis pour chaque étape. Après avoir décongelé les aliquotes, utilisez-les immédiatement. Ne pas recongeler. Suivez le tableau 5 pour un guide général de chaque type de support d’étape et des volumes nécessaires pour une plaque à 24 puits.
REMARQUE : Le moment optimal à chaque étape de différenciation peut varier selon la lignée cellulaire. Il est recommandé de vérifier l’expression de chaque marqueur cellulaire de stade pour améliorer l’efficacité de la différenciation. L’évaluation périodique de l’expression génique sur les cultures d’iPSCS n’a pas été incluse dans cette recherche car nous avons exploité des lots qualifiés par le fournisseur à de faibles passages, mais elle est recommandée comme étape de contrôle de la qualité pour les lignées d’iPSC nouvelles ou non encore qualifiées lors des étapes de différenciation ultérieures. - Stade 1 : Endoderme définitif (DE)
- Pour évaluer l’état de préparation à la différenciation, vérifiez les critères suivants.
- À l’aide d’un microscope, évaluez la confluence des colonies de cellules souches et le degré de différenciation spontanée dans la culture. Ne vous fiez pas uniquement à l’évaluation morphologique. La confluence optimale doit se situer entre 85 % et 90 % (au-dessus de ~105 cellules/cm2), et la différenciation spontanée minimale observée doit être de <5 % de différenciation.
- Prélever des cellules d’un puits sacrificiel pour la caractérisation par cytométrie en flux des marqueurs hiPSC avant de passer à l’étape suivante. Une stratégie de déclenchement par diffusion directe et latérale a été utilisée pour la caractérisation représentative de la cytométrie en flux. Si les cellules répondent aux critères optimaux, commencez la différenciation. Le critère de passage des cellules est >85% d’expression de 3 marqueurs idéalement (Tableau 4), en particulier en cas de nouvelles lignées iPSC / pas encore qualifiées en laboratoire.
REMARQUE : Les cultures d’iPSC cultivées sur différentes matrices subissent le même processus global avec seulement la croissance d’iPSC sur la matrice 4-XE, nécessitant des changements de milieu plus doux pour ne pas perturber les cellules semi-intégrées. Une mort cellulaire importante est attendue lors de l’induction de l’endoderme définitif, car les cellules sont particulièrement sensibles à ce stade. Faire preuve de prudence lors des changements de milieu, en visant à minimiser autant que possible la durée d’exposition des cellules à l’extérieur de l’environnement d’incubation à 37 °C.
- Commencer la différenciation des colonies cellulaires comme décrit ci-dessous.
- Jour 0 : Réchauffer une aliquote de DE à 37 °C. Pour éviter la dénaturation du milieu lors des cycles de réchauffement-refroidissement, assurez-vous de ne réchauffer que le volume requis pour le jour 0 (0,7 mL/puits).
- Aspirer les médias des hiPSC. Ajouter délicatement 0,7 ml de DE Medium par puits sur le côté du puits. Évitez le pipetage brutal qui pourrait détacher ou endommager les colonies. Incuber la plaque à 37 °C avec 5% de CO2 et 95% d’humidité pendant 24 h.
- Jour 1 : Réchauffer une aliquote de milieu DE contenant seulement le volume nécessaire pour le jour 1 (0,5 mL/puits) à 37 °C. Aspirez le milieu DE des cellules et ajoutez soigneusement 0,5 ml de milieu DE par puits sur le côté du puits. Incuber à 37 °C avec 5% de CO2 et 95% d’humidité pendant 24 h.
- Jour 3 : Les cellules sont prêtes à être analysées pour la formation définitive de l’endoderme. Reportez-vous au tableau 4 pour les marqueurs recommandés. Avant de passer à la phase suivante, sacrifiez un puits pour effectuer une caractérisation par cytométrie en flux de marqueurs spécifiques associés à l’ED. Une stratégie de déclenchement par diffusion directe et latérale a été utilisée pour la caractérisation représentative de la cytométrie en flux. Il est recommandé de vérifier l’expression de plus d'1 marqueur, en particulier dans le cas de lignes iPSC neuves/non encore qualifiées en laboratoire.
- Pour évaluer l’état de préparation à la différenciation, vérifiez les critères suivants.
- Stade 2 : Intestin moyen/hindgut (MH)
- Jour 3 : Réchauffer seulement le volume de milieu MH nécessaire pour le jour 3 (0,5 mL/puits) à 15 - 25 °C. Aspirer le milieu DE des cellules et le remplacer par 0,5 mL de milieu MH. Incuber à 37 °C avec 5% de CO2 et 95% d’humidité pendant 24 h.
- Jours 4 à 9 : Avant les changements quotidiens de milieu, utilisez le microscope à un grossissement de 2x ou 4x pour évaluer la formation de sphéroïdes en vérifiant la présence de structures 3D visibles (cela peut se produire à partir du jour 4) ; sphéroïdes flottants (peut survenir à partir du jour 5).
- Une fois que les sphéroïdes commencent à se détacher, à l’aide d’une pipette de 1 ml, transférez doucement le milieu des cellules dans un tube conique stérile de 15 ml afin d’évaluer le nombre de sphéroïdes détachés des cellules.
REMARQUE : Évitez toute force de cisaillement qui pourrait détacher les structures 3D de la monocouche ; Une fois prêts, les sphéroïdes se détacheront d’eux-mêmes. - Si moins de 50 sphéroïdes sont détachés, centrifuger à 200 x g pendant 5 min, retirer l’ancien milieu et remettre les sphéroïdes en suspension sur 0,5 mL/puits de milieu et transférer dans le puits respectif jusqu’à ce qu’un nombre suffisant de sphéroïdes aient mûri.
REMARQUE : La durée de l’exposition au milieu de l’intestin moyen et de l’intestin postérieur joue un rôle crucial dans la détermination de l’identité régionale des organoïdes de l’intestin grêle en développement, tels que le duodénum (exposition plus courte) ou l’iléon (exposition plus longue). Pour obtenir des cultures plus uniformes avec la même identité, essayez de collecter des sphéroïdes exposés simultanément à l’intestin moyen et à l’intestin postérieur. - Répétez l’étape 2.3.3 jusqu’à ce qu’un nombre suffisant de sphéroïdes soient détachés pour intégrer et amorcer la culture d’organoïdes intestinaux humains (hIO).
- Avant de passer à la phase suivante, séparez un échantillon de sphéroïdes et effectuez une caractérisation par cytométrie en flux de marqueurs spécifiques associés à l’intestin moyen/postérieur (MH). Une stratégie de déclenchement par diffusion directe et latérale a été utilisée pour la caractérisation représentative de la cytométrie en flux. Reportez-vous au tableau 4 pour une liste des marqueurs couramment utilisés. Il est recommandé de vérifier l’expression de plus d'1 marqueur, en particulier dans le cas de lignes iPSC neuves/non encore qualifiées en laboratoire.
- Étape 3 : Intégration du sphéroïde
- Préparez ce qui suit avant de commencer la procédure.
- Dégeler des aliquotes de Matrix 1-ABO et Matrix 3-ABO sur de la glace. Considérez le nombre de sphéroïdes collectés pour déterminer la quantité totale de matrice nécessaire si 30 à 40 μL de matrice sont nécessaires par dôme.
- Préparez 25 mL d’Advance DMEM/F-12 froid contenant 15 mM d’HEPES. Les ECM à haute concentration sont visqueuses et difficiles à pipeter. Placez une boîte d’embouts de pipette stériles de 100 μL à -20 °C pour préparer des pointes froides qui peuvent faciliter le processus. Préparez des aliquotes de milieu de croissance organoïde intestinal (OGM), qui nécessite 4 repas (0,5 ml/puits par repas). Voir le tableau 5 pour la préparation.
- Décongeler une aliquote du supplément OGM sur de la glace. Après avoir décongelé les aliquotes, utilisez-les immédiatement. Ne pas recongeler. Conserver l’OGM complet à une température comprise entre 2 et 8 °C pendant 2 semaines maximum.
- Préparez OGM contenant 3x du supplément par rapport à la formulation standard pour (3x OGM). Sortez le Matrix 4-XFO1 pensé Matrix 4-XFO4 du réfrigérateur et laissez-le se réchauffer à température ambiante (25 °C).
- Pour le système d’origine animale, effectuez les étapes décrites ci-dessous.
- Placez une plaque de culture tissulaire stérile à 24 puits dans l’incubateur pour qu’elle se réchauffe à 37 °C pendant la préparation des sphéroïdes et des matrices. Laissez les sphéroïdes collectés se déposer au fond d’un tube conique de 15 ml. Aspirez et jetez soigneusement le surnageant.
REMARQUE : Pour faire une comparaison plus précise de l’effet des matrices, assurez-vous de garder les sphéroïdes collectés à partir des hiPSC différenciés sur chaque matrice dans des tubes séparés. - Ajouter 1 mL de DMEM/F-12 avec 15 mM d’HEPES aux sphéroïdes. Centrifuger à 300 x g pendant 5 min à température ambiante (15 - 25 °C).
- Retirez avec précaution la plus grande partie possible du surnageant. Il est recommandé de commencer le prélèvement à l’aide d’une pipette de 1 mL et de passer à des pipettes de 100 μL et même de 10 μL pour prélever le plus possible sans déranger la pastille de sphéroïdes.
REMARQUE : Plus le volume de support restant est important, plus les matrices seront diluées. Les matrices diluées peuvent entraîner des problèmes de gélification ou des dômes plus mous. Des dômes plus mous augmentent les chances de s’effondrer avec un minimum de perturbations. - À l’aide d’une pipette avec une pointe de pipette froide de 100 μL, ajoutez 40 μL/50 sphéroïdes de Matrix 1-ABO ou Matrix 3-ABO froid (2 à 8 °C) dans leur tube respectif. Répartissez doucement les sphéroïdes dans la matrice en pipetant de haut en bas ~5x. Ne videz pas complètement l’embout de la pipette, ce qui pourrait introduire des bulles.
- Sortez la plaque de l’incubateur et, à l’aide d’une pointe de pipette froide, transférez doucement les sphéroïdes incrustés au centre de la plaque en suivant les étapes de la figure 2. Évitez de distribuer Matrix 1-ABO et Matrix 3-ABO trop rapidement dans la boîte de culture, car cela aplatirait le dôme.
- Transférez délicatement la plaque dans un incubateur à 37 °C et incubez pendant 30 min pour assurer la gélification du dôme. Faites particulièrement attention à toute secousse ou mouvement brusque pendant le transport et dans l’incubateur.
- Pendant l’incubation du dôme pour la gélification, réchauffer à 37 °C un volume suffisant d’OGM intestinal (0,5 mL/puits) pour le nombre de puits à utiliser.
REMARQUE : Réchauffez uniquement le volume nécessaire. Évitez les cycles de réchauffement-refroidissement. Assurez-vous que le support est chaud avant de l’ajouter au dôme ; Les fluides froids peuvent entraîner l’effondrement du dôme. - Après 30 min, ajoutez soigneusement 0,5 mL d’OGM intestinal sur le côté du puits pour ne pas déranger le dôme. Incuber à 37 °C avec 5% de CO2 et 95% d’humidité. Effectuez un changement complet de milieu tous les 3 à 4 jours en retirant le milieu, puis ajoutez du milieu frais.
- Placez une plaque de culture tissulaire stérile à 24 puits dans l’incubateur pour qu’elle se réchauffe à 37 °C pendant la préparation des sphéroïdes et des matrices. Laissez les sphéroïdes collectés se déposer au fond d’un tube conique de 15 ml. Aspirez et jetez soigneusement le surnageant.
- Pour les systèmes organoïdes Matrix 4-XF, suivez les étapes décrites.
REMARQUE : Le système organoïde Matrix 4-XF comprend quatre options différentes (O1-O4) ; La formulation de chaque type varie en termes de ligands biofonctionnels, de rigidité et de dégradabilité. Par conséquent, il est recommandé d’effectuer des expériences initiales pour déterminer le type optimal pour l’application spécifique. Ce protocole décrit l’utilisation des quatre options pour trouver une formulation optimale pour les organoïdes intestinaux dérivés des hiPSC. De plus, bien qu’il existe plusieurs protocoles de culture qui peuvent être utilisés (par exemple, l’encapsulation 3D, l’encapsulation en dôme), ce protocole implique l’utilisation de la méthode du dôme de sorte qu’il est possible de comparer directement avec les méthodes courantes utilisées lors du travail avec des systèmes de membrane basale d’origine animale.- Ajouter 1 mL/puits de DMEM/F-12 d’avance à froid contenant 16 mM d’HEPES aux sphéroïdes générés sur le système Matrix 4-XFO4. Centrifuger à 300 x g pendant 5 min à température ambiante (15 - 25 °C).
REMARQUE : Pour trouver la formulation optimale pour l’application, assurez-vous de répartir les sphéroïdes dans 4 tubes séparés avant la centrifugation. Les sphéroïdes générés sur des matrices d’origine animale peuvent également être transformés en organoïdes intestinaux à l’aide de ce système. - À l’aide d’une pipette de 1 ml, prélever le surnageant sans déranger les sphéroïdes. Ajouter 50 μL de 3x OGM à la pastille de sphéroïdes (~100 sphéroïdes). Ajoutez 100 μL de la matrice 4-XFO sélectionnée à la suspension sphéroïde de 50 μL et mélangez doucement 5 à 10 fois plus. Conservez la matrice 4-XFO à 3x OGM à un rapport de mélange v/v de 2:1 pour avoir une concentration finale de 1x.
- Ajouter 40 μL du mélange hydrogel-sphéroïde au centre d’une plaque de culture tissulaire à 24 puits. Transférez délicatement les plaques dans un incubateur à 37 °C et incubez pendant 30 min.
REMARQUE : Matrix 4-XFO ne nécessite pas d’incubation à 37 °C pour la gélification ; cependant, il est recommandé de garder les organoïdes exposés à des conditions similaires par rapport aux systèmes Matrix 1-ABO et Matrix 3-ABO qui nécessitent 37 °C pour une meilleure gélification des dômes. - Après 30 min, ajoutez soigneusement 0,5 mL d’OGM intestinal sur le côté du puits afin de ne pas perturber le dôme. Incuber à 37 °C avec 5% de CO2 et 95% d’humidité. Effectuez un changement complet de milieu tous les 3 à 4 jours en retirant le milieu, puis en ajoutant du milieu frais.
- Ajouter 1 mL/puits de DMEM/F-12 d’avance à froid contenant 16 mM d’HEPES aux sphéroïdes générés sur le système Matrix 4-XFO4. Centrifuger à 300 x g pendant 5 min à température ambiante (15 - 25 °C).
- Préparez ce qui suit avant de commencer la procédure.
- Passage et maturation hIO
- Préparez les mêmes produits chimiques, solutions et réactifs qu’à l’étape 2.4.1.
- Ajouter 1 à 2 ml de solution de rinçage anti-adhérence dans un tube conique de 15 ml (1 par condition) et agiter pour enrober le tube.
- Retirer la solution anti-adhérence et rincer les tubes avec 5 mL de D-PBS (sans Ca++ et Mg++). Boucher tous les tubes revêtus et les conserver à température ambiante (15 - 25 °C) jusqu’à ce que vous en ayez besoin.
- Aspirez le fluide à partir des dômes. À l’aide d’une pipette de 1 ml, ajoutez 1 mL de DMEM/F-12 froid directement dans le dôme. Le but est de détacher les dômes de la plaque.
- Ajoutez 1 ml supplémentaire de DMEM/F-12 froid dans le puits et pipetez de haut en bas pour récolter les organoïdes restants. Transférer dans les tubes coniques enrobés de 15 mL.
REMARQUE : Vérifiez la récolte réussie des organoïdes en examinant visuellement le puits au microscope. Si des organoïdes résiduels sont observés, répétez l’étape 2.5.5. - À l’aide d’un pipeteur de 1 ml, effectuer un pipetage ascendant et descendant de la suspension pour désintégrer les organoïdes jusqu’à ce qu’une suspension de fragments uniforme soit obtenue avec la taille d’organoïde souhaitée (p. ex., 100 à 500 μm).
REMARQUE : Utilisez une pipette de 200 μL pour vérifier que les organoïdes sont conformes à la taille recommandée. L’utilisation de la pipette de 200 μL facilite une fragmentation supplémentaire si nécessaire, garantissant que les fragments peuvent passer en douceur à travers une pointe de pipette de 200 μL.
ATTENTION : Évitez de briser des fragments en cellules uniques par un pipetage brutal ou prolongé. - Établissez la densité organoïde souhaitée en comptant les fragments ou en utilisant le rapport de division. Séparer une aliquote et effectuer le comptage en utilisant la même procédure que celle décrite aux étapes 1.4.6 à 1.4.9 pour le comptage des grumeaux.
REMARQUE : La densité optimale des organoïdes doit être optimisée par ligne ; En général, une densité de 40 à 80 organoïdes intestinaux par dôme est recommandée. - Assurez-vous que le tube est placé sur de la glace pendant le processus de comptage des organoïdes. Après environ 5 min, des fragments d’organoïdes se seront déposés au fond du tube en raison de la gravité.
REMARQUE : Plus le volume de la solution organoïde est important, plus il faudra de temps pour se déposer au fond. - Éliminez doucement la quantité de surnageant et la couche trouble formée sur les organoïdes. Dans les premiers passages où les organoïdes mûrissent, cette phase trouble englobe une matrice et des cellules individuelles.
- Ajouter 2 mL de DMEM/F-12 froid en pipetant directement sur la pastille. Centrifuger à 200 x g pendant 5 min à température ambiante (15 - 25 °C).
- Dans le cas d’un système d’origine animale, retirer et jeter avec précaution le surnageant en suivant les mêmes étapes que celles décrites aux étapes 2.4.2.4 à 2.4.2.9.
- Pour le système Matrix 4-XFO, retirez et jetez soigneusement le surnageant en suivant les mêmes étapes que celles décrites aux étapes 2.4.3.2-2.4.3.4.
REMARQUE : Lorsque vous travaillez avec le système Matrix 4-XFO, il est recommandé d’utiliser une solution de récupération d’organoïdes d’hydrogel sans xéno pour une élimination optimale des résidus d’hydrogel. Cette solution de récupération est particulièrement recommandée lors du passage d’un système d’hydrogel d’origine animale à un système sans xénogène afin de garantir l’élimination de toute matière xénogénique.

Graphique 2. Schéma de la technique recommandée pour la formation du dôme. Le schéma décrit le processus étape par étape recommandé pour une formation réussie du dôme pour tous les systèmes. Veuillez cliquer ici pour voir une version agrandie de cette figure.
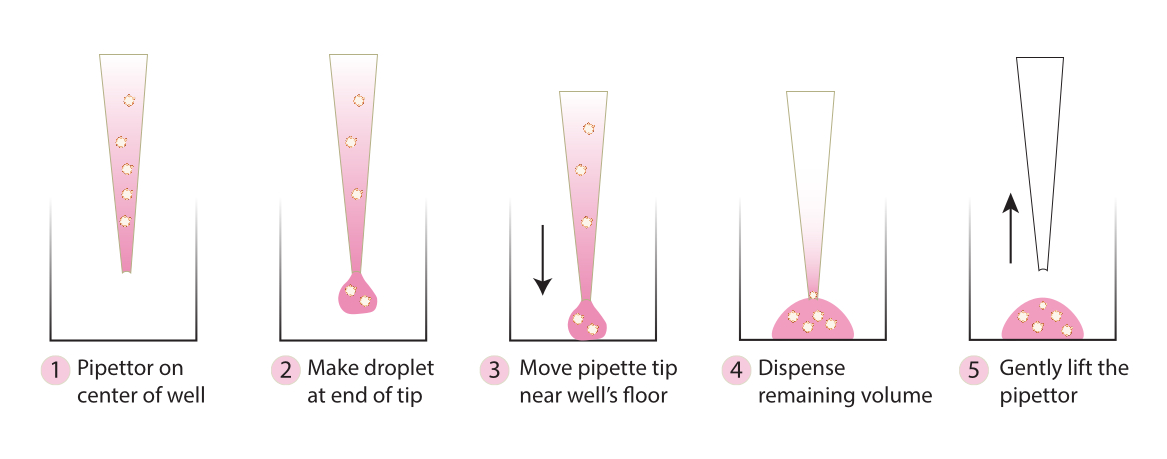{kind=link}
3. Caractérisation de la taille des E/S
REMARQUE : La taille des organoïdes a été caractérisée par des images en fond clair prises à 4x et 10x. L’analyse du traitement d’image a été automatisée à l’aide de MATLAB. Les étapes générales du processus sont décrites ci-dessous, et un échantillon du code est inclus dans le fichier supplémentaire 1.
- Définissez le répertoire contenant les images et listez tous les fichiers image qu’il contient. Initialisez la table pour stocker les résultats. Lisez les images en fond clair dans un fichier.
- Définissez la conversion du pixel en μm et réglez l’échelle. Si l’image ne contient pas de barre d’échelle, demandez à l’utilisateur le facteur de conversion pixel-μm.
- Convertissez l’image en niveaux de gris. Appliquez un filtre gaussien pour réduire le bruit dans l’image. Seuilz l’image filtrée pour séparer les organoïdes de l’arrière-plan.
- Remplissez les petits trous et retirez les petits objets pour nettoyer l’image binaire. Effectuez une analyse de composants connectés pour identifier les organoïdes individuels et calculer leurs propriétés telles que l’aire, le centroïde, la longueur du grand axe et la longueur du petit axe.
- Calculez la taille de l’organoïde en fonction des longueurs des axes principal et mineur. Affichez les organoïdes segmentés sur l’image d’origine et étiquetez-les avec leur taille. Imprimez les tailles des organoïdes et stockez les résultats dans un fichier .cvs.
REMARQUE : Les deux premières images ont été analysées manuellement afin de déterminer les paramètres optimaux pour l’écart-type du filtre gaussien et la zone jusqu’au seuil. Le code du fichier supplémentaire 1 fournit un exemple du cadre de base pour l’analyse de la taille des organoïdes à partir d’images en fond clair ; Cependant, des améliorations supplémentaires sont nécessaires pour répondre aux exigences spécifiques de chaque type et qualité d’image. Le même processus peut être effectué à l’aide d’un logiciel open source comme FIJI d’Image J. - Suivez les étapes ci-dessous pour utiliser le logiciel FIJI.
- Définissez la conversion du pixel en μm et réglez l’échelle. Si l’image ne contient pas de barre d’échelle, demandez à l’utilisateur le facteur de conversion pixel-μm en cliquant sur Analyser > Définir l’échelle.
- Convertissez l’image en niveaux de gris en cliquant sur Image > Type > 8 bits. Appliquez un filtre gaussien pour réduire le bruit dans l’image en cliquant sur Traiter > Filtre > Texte de présentation gaussien > Sigma (rayon) utilisé : 2.
- Limitez l’image filtrée pour séparer les organoïdes de l’arrière-plan en cliquant sur Image > Seuil > MaxEntropy > Appliquer.
- Remplissez les petits trous et supprimez les petits objets pour nettoyer l’image binaire en cliquant sur Traiter > bruit > Supprimer les valeurs aberrantes > 20 pixels.
- Effectuez une analyse de composants connectés pour identifier les organoïdes individuels et calculer leurs propriétés telles que l’aire, le centroïde, la longueur de l’axe principal et la longueur de l’axe mineur en cliquant sur Analyser > Définir la mesure > Assurez-vous que l’aire, le périmètre et le diamètre sont inclus > Analyser > Analyser les particules > Afficher les contours.
- Analyse statistique
- Évaluez la normalité de la distribution des données à l’aide du test Saphiro-Wilk à l’aide du logiciel JMP (SAS). Pour examiner les différences statistiques entre les groupes, effectuez une ANOVA à deux facteurs et effectuez des tests post-hoc à l’aide de la méthode de Wilcoxon non paramétrique dans le logiciel JMP(SAS). La signification a été établie à un niveau alpha de p ≤ 0,05.
Résultats
Selon ce protocole, des membranes basales disponibles dans le commerce et un système d’hydrogel sans xénogène ont été utilisés avec succès pour cultiver des cellules hiPSC et les différencier en hIO. L’objectif principal de ces expériences était d’évaluer systématiquement l’équivalence de matrices provenant de diverses sources pour les travaux hiPSC et hIO. La première section de ce protocole s’est concentrée sur le maintien et la caractérisation d’une culture d’iPSC saine qui produit une g?...
Discussion
La sélection du microenvironnement optimal pour le travail sur les cellules souches et les organoïdes est une première étape cruciale lors de l’utilisation de ces plateformes pour un large éventail d’applications. Nos résultats représentatifs montrent que la matrice 4-XFO3, en combinaison avec une concentration plus élevée de facteurs de croissance, conduit à des organoïdes plus grands, ce qui suggère que les propriétés physiques des hydrogels sans xénogrammes peuvent être exploitées pour optimiser l...
Déclarations de divulgation
Le Dr John Huang est fondateur et PDG de TheWell Bioscience.
Remerciements
Les auteurs reconnaissent la formation antérieure et les recommandations générales concernant le début du travail sur les hiPSC et les organoïdes des Drs Christina Pacak, Silveli Susuki-Hatano et Russell D’Souza. Ils remercient la Dre Chelsey Simmons pour ses conseils dans l’utilisation des systèmes d’hydrogel pour les travaux de culture cellulaire in vitro . De plus, les auteurs tiennent à remercier les Drs Christine Rodriguez et Thomas Allison de STEMCELL Technologies pour leurs conseils sur la culture hiPSC. Les auteurs remercient également TheWell Bioscience d’avoir couvert les frais de publication.
matériels
| Name | Company | Catalog Number | Comments |
| 24-Well Plate (Culture treated, sterile) | Falcon | 353504 | |
| 37 °C water bath | VWR | ||
| 96-well plate | Fisher Scientific | FB012931 | |
| Advanced DMEM/F12 | Life Technologies | 12634 | |
| Anti-adherence Rinsing Solutio | STEMCELL Technologies | 7010 | |
| Biological safety cabinet (BSC) | Labconco | Logic | |
| Brightfield Microscope | Echo Rebel | REB-01-E2 | |
| BXS0116 | ATCC | ACS-1030 | |
| Centrifuge with temperature control (4 °C capabilities) | ThermoScientific | 75002441 | |
| Conical tubes, 15 mL, sterile | Thermo Fisher Scientific | 339650 | |
| Conical tubes, 50 mL, sterile | Thermo Fisher Scientific | 339652 | |
| Cultrex RGF BME, Type 2 | Bio-techne | 3533-005-02 | |
| Cultrex Stem Cell Qualified RGF BME | Bio-techne | 3434-010-02 | |
| D-PBS (Without Ca++ and Mg++) | Thermo Fisher Scientific | 14190144 | |
| GeltrexLDEV-Free, hESC-Qualified Reduce Growth Factor | Gibco | A14133-02 | |
| GlutaMAX Supplement | Thermo Fischer Scientific | 35050-061 | |
| Guava Muse Cell Analyzer or another flow cytometry equipment (optional) | Luminex | 0500-3115 | |
| HEPES buffer solution | Thermo Fischer Scientific | 15630-056 | |
| Heralcell Vios Cell culture incubator (37 °C, 5% CO2) | Thermo Scientific | 51033775 | |
| JMP Software | SAS Institute | JMP 16 | |
| MATLAB | MathWorks, Inc | R2022b | |
| Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix LDEV free | Corning | 356231 | |
| Matrigel Matrix High Concentration (HC), Growth Factor Reduced (GFR) LDEV-free | Corning | 354263 | |
| mTeSR Plus Medium | STEMCELL Technologies | 100-0276 | |
| Nunclon Delta surface treated 24-well plate | Thermo Scientific | 144530 | |
| PE Mouse Anti-human CD326 (EpCAM) | BD Pharmingen | 566841 | |
| PE Mouse Anti-human CDX2 | BD Pharmingen | 563428 | |
| PE Mouse Anti-human FOXA2 | BD Pharmingen | 561589 | |
| PerCP-Cy 5.5 Mouse Anti-human SSEA4 | BD Pharmingen | 561565 | |
| ReLeSR | STEMCELL | 5872 | |
| SCTi003-A | STEMCELL Technologies | 200-0510 | |
| Serological pipettes (10 mL) | Fisher Scientific | 13-678-11E | |
| Serological pipettes (5 mL) | Fisher Scientific | 13-678-11D | |
| STEMdiff Intestinal Organoid Growth Medium | STEMCELL Technologies | 5145 | |
| STEMdiff Intestinal Organoid Kit | STEMCELL Technologies | 5140 | |
| Vitrogel Hydrogel Matrix | TheWell Bioscience | VHM01 | |
| VitroGel ORGANOID Discovery Kit | TheWell Bioscience | VHM04-K |
Références
- Hynes, R. O. Integrins: Bidirectional, allosteric signaling machines. Cell. 110 (6), 673-687 (2002).
- Frantz, C., Stewart, K. M., Weaver, V. M. The extracellular matrix at a glance. J Cell Sci. 123, 4195-4200 (2010).
- Hinz, B., Gabbiani, G. Fibrosis: Recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol Rep. 2, 78 (2010).
- Pickup, M. W., Mouw, J. K., Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15 (12), 1243-1253 (2014).
- Rozario, T., DeSimone, D. W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev Biol. 341 (1), 126-140 (2010).
- Even-Ram, S., Artym, V., Yamada, K. M. Matrix control of stem cell fate. Cell. 126 (4), 645-647 (2006).
- Engler, A. J., Sen, S., Sweeney, H. L., Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell. 126 (4), 677-689 (2006).
- Tran, O. N., et al. Organ-specific extracellular matrix directs trans-differentiation of mesenchymal stem cells and formation of salivary gland-like organoids in vivo. Stem Cell Res Ther. 13 (1), 306 (2022).
- Nikolaev, M., et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature. 585 (7826), 574-578 (2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., et al. Tissue geometry drives deterministic organoid patterning. Science. 375 (6576), (2022).
- Heo, J. H., Kang, D., Seo, S. J., Jin, Y. Engineering the extracellular matrix for organoid culture. Int J Stem Cells. 15 (1), 60-69 (2022).
- Shamir, E. R., Ewald, A. J. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 15 (10), 647-664 (2014).
- Clevers, H. Modeling development and disease with organoids. Cell. 165 (7), 1586-1597 (2016).
- Jung, P., et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 17 (10), 1225-1227 (2011).
- Lancaster, M. A., Knoblich, J. A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science. 345 (6194), 1247125 (2014).
- Huch, M., et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 494 (7436), 247-250 (2013).
- Greenlee, A. R., Kronenwetter-Koepel, T. A., Kaiser, S. J., Liu, K. Comparison of Matrigel and gelatin substrata for feeder-free culture of undifferentiated mouse embryonic stem cells for toxicity testing. Toxicol In Vitro. 19 (3), 389-397 (2005).
- Geltrex LDEV-Free, HESC-Qualified, Reduced Growth Factor Basement Membrane Matrix User Guide (Pub.No. MAN0007336 3.0. Fisher Scientific Available from: https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.cn/TFS-Assets%2FLSG%2Fmanuals%2FGeltrex_LDEV_Free_hESC_qualified_PI.pdf (2024)
- biotechne R&D Systems. Cultrex Stem Cell Qualified Reduced Growth Factor. biotechne R&D Systems. , (2024).
- VitroGel Organoid Protocol. TheWell Bioscience Available from: https://www.thewellbio.com/video-protocols (2024)
- Spence, J. R., et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 470 (7332), 105-110 (2011).
- Henderson, J. K., et al. Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells. 20 (4), 329-337 (2002).
- Haruna, N. F., Huang, J. Investigating the dynamic biophysical properties of a tunable hydrogel for 3D cell culture. J Cytol Tissue Biol. 7, 30 (2020).
- Cherne, M. D., et al. A synthetic hydrogel, VitroGel ORGANOID-3, improves immune cell-epithelial interactions in a tissue chip co-culture model of human gastric organoids and dendritic cells. Front Pharmacol. 12, 707891 (2021).
- Stewart, D. C., et al. Quantitative assessment of intestinal stiffness and associations with fibrosis in human inflammatory bowel disease. PLoS One. 13, e0200377 (2018).
- Hernandez-Gordillo, V., et al. Fully synthetic matrices for in vitro culture of primary human intestinal enteroids and endometrial organoids. Biomaterials. 254, 120125 (2020).
- Broguiere, N., et al. Growth of epithelial organoids in a defined hydrogel. Adv Mater. 30, 1801621 (2018).
- Barthes, J., et al. Cell microenvironment engineering and monitoring for tissue engineering and regenerative medicine: The recent advances. BioMed Res Int. 2014, 921905 (2014).
- Engler, A. J., Sen, S., Sweeney, H. L., Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell. 126 (4), 677-689 (2006).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nat Rev Mater. 5 (7), 539-551 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.