Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Messung dynamischer glykosomaler pH-Änderungen bei lebenden Trypanosoma brucei
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Wir beschreiben eine Methode, um zu untersuchen, wie der pH-Wert auf Umweltreize in den Glykosomen der Blutkreislaufform afrikanischer Trypanosomen reagiert. Dieser Ansatz beinhaltet einen pH-sensitiven Sensor für vererbbare Proteine in Kombination mit Durchflusszytometrie zur Messung der pH-Dynamik, sowohl als Zeitverlaufsassay als auch in einem Hochdurchsatz-Screen-Format.
Zusammenfassung
Der Glukosestoffwechsel ist für das afrikanische Trypanosom, Trypanosoma brucei, als essentieller Stoffwechselprozess und Regulator der Parasitenentwicklung von entscheidender Bedeutung. Über die zellulären Reaktionen, die entstehen, wenn sich der Glukosespiegel in der Umwelt ändert, ist wenig bekannt. Sowohl in Parasiten im Blutkreislauf als auch in prozyklischer Form (Insektenstadium) beherbergen Glykosomen den größten Teil der Glykolyse. Diese Organellen werden als Reaktion auf Glukoseentzug schnell angesäuert, was wahrscheinlich zu einer allosterischen Regulation von glykolytischen Enzymen wie Hexokinase führt. In früheren Arbeiten war die Lokalisierung der chemischen Sonde, die zur Durchführung von pH-Messungen verwendet wird, eine Herausforderung, was ihren Nutzen in anderen Anwendungen einschränkte.
Diese Arbeit beschreibt die Entwicklung und Verwendung von Parasiten, die glykosomal lokalisiertes pHluorin2, einen vererbbaren Protein-pH-Biosensor, exprimieren. pHluorin2 ist eine ratiometrische pHluorin-Variante, die eine pH-abhängige (säureabhängige) Abnahme der Anregung bei 395 nm zeigt, während sie gleichzeitig eine Zunahme der Anregung bei 475 nm ergibt. Transgene Parasiten wurden durch Klonierung des offenen pHluorin2-Leserahmens in den Trypanosomen-Expressionsvektor pLEW100v5 erzeugt, was eine induzierbare Proteinexpression in beiden Lebenszyklusstadien ermöglicht. Immunfluoreszenz wurde verwendet, um die glykosomale Lokalisation des pHluorin2-Biosensors zu bestätigen, indem die Lokalisation des Biosensors mit der glykosomalen residenten Proteinaldolase verglichen wurde. Die Reaktionsfähigkeit des Sensors wurde bei unterschiedlichen pH-Werten kalibriert, indem Zellen in einer Reihe von Puffern mit einem pH-Wert von 4 bis 8 inkubiert wurden, ein Ansatz, den wir zuvor zur Kalibrierung eines pH-Sensors auf Fluoresceinbasis verwendet haben. Anschließend maßen wir die pHluorin2-Fluoreszenz bei 405 nm und 488 nm mittels Durchflusszytometrie zur Bestimmung des glykosomalen pH-Werts. Wir validierten die Leistung der lebenden transgenen pHluorin2-exprimierenden Parasiten und überwachten den pH-Wert im Laufe der Zeit als Reaktion auf Glukoseentzug, einen bekannten Auslöser der glykosomalen Ansäuerung bei PF-Parasiten. Dieses Tool hat eine Reihe potenzieller Anwendungen, einschließlich des potenziellen Einsatzes im Hochdurchsatz-Wirkstoffscreening. Über den glykosomalen pH-Wert hinaus könnte der Sensor an andere Organellen angepasst oder in anderen Trypanosomatiden verwendet werden, um die pH-Dynamik in der lebenden Zellumgebung zu verstehen.
Einleitung
Parasitäre Kinetoplastiden sind wie die meisten lebenden Organismen auf Glukose als grundlegenden Bestandteil des zentralen Kohlenstoffstoffwechsels angewiesen. Zu dieser Gruppe gehören medizinisch wichtige Organismen wie das afrikanische Trypanosom Trypanosoma brucei; das amerikanische Trypanosom, T. cruzi; und Parasiten der Gattung Leishmania. Der Glukosestoffwechsel ist entscheidend für das Wachstum von Parasiten in den pathogenen Lebenszyklusstadien. Wenn beispielsweise Glukose entzogen wird, stirbt die Blutbahnform (BSF) des afrikanischen Trypanosoms schnell ab. Insbesondere dient die Glykolyse als einzige ATP-Quelle in diesem Stadium der Infektion1. Leishmanienparasiten sind ebenfalls von Glukose im menschlichen Wirt abhängig, wobei das Amastigoten-Lebenszyklusstadium, das sich in Wirtsmakrophagen befindet, für das Wachstum auf diese Kohlenstoffquelle angewiesenist 2.
Während diese Parasiten unterschiedliche Lebensstile mit unterschiedlichen Insektenvektoren haben, haben sie viele Gemeinsamkeiten in der Art und Weise, wie sie auf Glukose reagieren und diese konsumieren. Zum Beispiel lokalisieren diese Parasiten die meisten glykolytischen Enzyme in modifizierten Peroxisomen, die Glykosomen genannt werden. Diese Kinetoplastid-spezifische Organelle ist mit Säugetier-Peroxisomen verwandt, basierend auf konservierten Biosynthesemechanismen und Morphologie 3,4,5,6.
Die Kompartimentierung der meisten Enzyme des glykolytischen Signalwegs in das Glykosom bietet parasitenspezifische Mittel zur Regulierung des Signalwegs. Mit Hilfe einer chemischen pH-Sonde konnten wir feststellen, dass Nährstoffentzug eine schnelle Ansäuerung von Parasitenglykosomen in prozyklischer Form (PF) auslöst, die zu einer veränderten glykolytischen Enzymaktivität führt, indem eine allosterische Regulatorbindungsstelle auf dem glykolytischen Schlüsselenzym Hexokinaseexponiert wird 7,8. In unseren früheren Arbeiten musste die chemische Sonde für den Einsatz ständig abgegeben werden, was ihren Nutzen in anderen Anwendungen einschränkte. Darüber hinaus schränkten Herausforderungen bei der Aufrechterhaltung der Sondenverteilung im Glykosom im BSF den Nutzen des Ansatzes zur Untersuchung des glykosomalen pH-Werts in diesem Lebensstadium ein.
In dieser Studie haben wir den fluoreszierenden Protein-Biosensor pHluorin2 verwendet, um die glykosomale pH-Änderung in BSF T. brucei als Reaktion auf Umweltreize wie Glukosemangelzu überwachen 9 (Abbildung 1). Die Ergebnisse dieser Arbeit deuten darauf hin, dass BSF T. brucei Glykosomen als Reaktion auf Hunger schnell reversibel ansäuert, ähnlich wie wir es bei PF-Parasiten beobachtet haben. Wir erwarten, dass dieser Biosensor unser Verständnis der glykolytischen Regulation in T. brucei und verwandten Parasiten verbessern wird.
Protokoll
Die Verwendung von T. brucei brucei 90-13 BSF-Trypanosomen, einer monomorphen Parasitenlinie, erfordert eine Berücksichtigung der Sicherheit, da sie als Organismen der Risikogruppe 2 gelten, die in Einrichtungen der Biosicherheitsstufe 2 gehandhabt werden sollten.
1. Trypanosomenkultur und Transfektion
- Kultur T. brucei brucei 90-13 BSF-Trypanosomen in HMI-9-Medium, ergänzt mit 10 % hitzeinaktiviertem FBS und 10 % Nu-Serum bei 37 °C in 5 % CO210.
HINWEIS: Um die Kultur gesund zu halten, halten Sie die Zelldichte zwischen 2 × 104 und 5 × 106 Zellen/ml. - Klonierung von pHluorin2-PTS1 in pLEW100v5
- Synthetisieren Sie den offenen pHluorin2-Leserahmen kommerziell, um das Gen mit einer 3'-Erweiterung zu erhalten, die für einen Zwei-Glycin-Linker kodiert, gefolgt von dem Tripeptid-AKL, einem PTS1-Lokalisierungstag.
- Klonen Sie dieses Konstrukt durch Restriktionsverdau in den induzierbaren Trypanosomenexpressionsvektor pLEW100v5. Verdauen Sie sowohl den Klonierungsvektor, der pHluorin2-PTS1 als auch pLEW100v5 enthält, mit HindIII und BamHI11. Führen Sie einen Reinigungsschritt durch, vorzugsweise durch Agarosegelreinigung, um Restriktionsenzyme, das unerwünschte Klonierungsvektor-Rückgrat und das herausgeschnittene Luciferase-Gen-enthaltende Fragment zu entfernen.
- Lilaminieren Sie den pHluorin2-haltigen Einsatz in verdautes pLEW100v5 mit T4-DNA-Ligase12. (siehe Ergänzende Abbildung S1 für ein Schema zum Klonen).
- Sequenzieren Sie das Plasmid durch Ganzplasmidsequenzierung der nächsten Generation, um zu überprüfen, ob das Insert und der Vektor korrekt ligiert sind und dass während des Klonierungsprozesses keine Mutationen im pHluorin2-PTS1-Gen eingeführt werden.
HINWEIS: Die vollständige Plasmidsequenz wurde an Addgene.org übermittelt und erhielt die Zuweisung #83680. - Linearisierung von 20 μg des Plasmids durch Verdau mit 40 Einheiten NotI; dann transfizieren Sie in BSF 90-13-Parasiten durch Nukleofektion mit dem referenzierten humanen T-Zell-Kit (siehe Materialtabelle). Wählen Sie für eine stabile Integration, wie von Burkard et al.13 beschrieben.
2. Immunfluoreszenz-Kolokalisation von pHlourin2-PTS1
- Mikroskopie-Objektträger vorbereiten, indem sie 10 Minuten lang mit Poly-L-Lysin 0,1 % (w/v) in H2O behandelt werden. Waschen Sie den Objektträger nach dem Entfernen der Poly-L-Lysin-Lösung einmal mit PBS.
- Um die pHluorin2-PTS1-Expression zu induzieren, behandeln Sie die Zellen 24 Stunden vor der Ernte bei 2 × 105 Zellen/ml mit Tetracyclin oder Doxycyclin (1 μg/ml). Pellet 2 × 106 elterliche und induzierte pLEWpHluorin2-PTS1 (pHL)-Parasiten durch Zentrifugation (Raumtemperatur [RT], 10 min, 1.000 × g) und einmal mit PBS waschen.
- Resuspendieren Sie die Zellen in 200 μl frisch zubereitetem 2%igem Paraformaldehyd in PBS, hergestellt aus handelsüblicher 16%iger EM-Lösung. Tragen Sie die Zellen im Fixiermittel auf den Objektträger auf und lassen Sie die Zellen 30 Minuten lang ruhen. Waschen Sie die anhaftenden Zellen 2x mit Waschlösung (0,1% normales Ziegenserum in PBS).
- Die Permeabilisierungslösung (0,5% Triton X-100 in PBS) auf die Zellen auftragen und genau 30 Minuten inkubieren. Entfernen Sie die Permeabilisierungslösung und waschen Sie sie einmal mit reichlich Waschlösung.
- Die Blocklösung (10% normales Ziegenserum und 0,1% Triton X-100 in PBS) auftragen und 30 Minuten inkubieren.
- Die gegen T. brucei Aldolase erhobenen Antiseren werden 1:500 in Blocklösung verdünnt und auf die Zellen14 aufgetragen. 1 h bei RT inkubieren. Waschen Sie die Objektträger 5x für 3-5 min min mit Waschlösung.
- Ziegen-Anti-Kaninchen-Alexa Fluor 568 1:1.000 in Blocklösung verdünnen und auf die Zellen auftragen. 1 h bei RT inkubieren. Waschen Sie die Objektträger 5x für 3-5 min mit Waschlösung.
- Tragen Sie Montagemedium auf und versiegeln Sie ein Deckglas auf dem Objektträger.
- Bilden Sie die Zellen mit einem 100x-Objektiv (NA 1.4-0.7) ab und analysieren Sie die Bilder mit ImageJ. Führen Sie die Kolokalisierungsanalyse von Pearson mit dem Plug-in "Coloc 2" für ImageJ durch. Siehe Ergänzende Abbildung S2 für ein Feld repräsentativer Zellen. )
- Fügen Sie dazu die Bilddatei zu Fiji hinzu und wählen Sie Bilder aus.
- Passen Sie Helligkeit und Kontrast für jeden Kanal bis zu einem Punkt an, an dem kein Hintergrund sichtbar ist.
- Ändern Sie Bilder von 16-Bit in 8-Bit, führen Sie die Bilder zusammen und schneiden Sie sie dann auf eine einzelne Zelle mit geteilten Kanälen zu.
- Wählen Sie unter Analyse und Kolokalisierung das Plug-in Coloc2 aus. Wählen Sie aldolase (der rote Kanal) als Kanal 1 und pHL (der grüne Kanal) als Kanal 2 aus, und klicken Sie auf OK , um die Berechnung der Pearson-Korrelation zu starten.
3. Probenvorbereitung für die Durchflusszytometrie
- Induzieren Sie pHL-Zellen über Nacht entweder mit Tetracyclin oder Doxycyclin (1 μg/ml).
- Pelletieren Sie die Zellen (~4 × 107 pHL und ~1 × 106 parental) durch Zentrifugation (RT, 10 min, 1.000 × g). Entfernen Sie den Überstand und resuspendieren Sie die Zellen in 1 ml PBS entweder mit oder ohne 10 mM Glukose, je nachdem, ob die Probe ausgehungert oder nicht ausgehungert ist. Für Zeitverlaufstests resuspendieren Sie in PBS, das mit 10 mM Glukose ergänzt wird, um zu verhindern, dass die Zellen verhungern, bis die Waschungen abgeschlossen sind. Für den Hochdurchsatz-Screening-Assay (HTS) resuspendieren Sie in PBS ohne Glukose, um die Verschleppung von Glukose zu minimieren. Wiederholen Sie die Wäschen noch zweimal.
- Zentrifugieren Sie die Zellen ein viertes Mal, entfernen Sie den Überstand und resuspendieren Sie das Zellpellet je nach Behandlung entweder in PBS, PBS plus 5 mM Glukose oder PBS plus 10 mM Glukose. Ergänzen Sie die Proben entweder mit 1 μg/ml Propidiumiodid (PI) oder 100 nM Thiazolrot (TR) zur Lebend-/Totbestimmung. Übertragen Sie jede Probe in 5-ml-Röhrchen, die mit dem Durchflusszytometer kompatibel sind.
4. Durchflusszytometrie
HINWEIS: Bereiten Sie das Experiment auf einem Durchflusszytometer vor, das die folgenden Laser enthält: 405 nm (violett), 488 nm (blau) und 561 nm (gelb) oder 638 nm (rot). Siehe Ergänzende Tabelle S1 für gebräuchliche Namen für Kanäle, die unten besprochen werden.
- Zur Messung der pHL-Fluoreszenz verwenden Sie die Kanäle KO525 (VL2-H, Anregung 405 nm, Emission 542/27 nm) und FITC (BL1-H, Anregung 488 nm, Emission 530/30 nm). Um lebende Zellen von toten Zellen zu unterscheiden, verwenden Sie entweder PI oder TR; PI auf dem YL2-H-Kanal messen (Anregung 561 nm, Emission 620/25 nm). Messen Sie TR auf dem RL1-H-Kanal (638 nm Anregung, 660/10 BP).
- Um das Experiment in der Durchflusszytometer-Software einzurichten, erstellen Sie die folgenden Diagramme: 1) YL2-H-Kanalhistogramm (bei Verwendung von PI) oder ein RL1-H-Kanalhistogramm (bei Verwendung von TR), 2) Punktdiagramm von FSC-A vs. SSC-A, 3) Punktdiagramm von FSC-A vs. FSC-H und 4) Punktdiagramm von BL1-H vs. VL2-H.
- Setzen Sie zuerst die ungefärbte WT-Steuerung (Parental Cell-Line) auf den Probeninjektionsanschluss (SIP) und heben Sie den Tisch in Position. Beginnen Sie mit der Erfassung von Daten auf dem Zytometer mit der niedrigsten Durchflussrate, um Zeit für die erforderlichen Anpassungen zu haben. Um zu vermeiden, dass falsche Ablagerungen und tote Zellen entstehen, beginnen Sie 5 s nach Beginn der Probenaufnahme mit der Aufzeichnung von Ereignissen.
- Stellen Sie die YL2- oder RL1-Spannung so ein, dass der Hauptpeak innerhalb von 103-10 4 relativen Fluoreszenzintensitätseinheiten (RFI) liegt. Passen Sie die FSC- und SSC-Spannungen so an, dass >90 % der Ereignisse in das Punktdiagramm passen. Passen Sie den FSC-Schwellenwert an, um die Trümmerpopulation, aber keine wahrscheinlichen Zellen auszuschließen.
- Stellen Sie die VL2- und BL1-Kanäle so ein, dass der primäre Peak innerhalb von 103-10 4 RFI-Einheiten für die nicht gefärbte WT-Steuerung liegt.
- Legen Sie die erste Probe mit induziertem pHL, die entweder mit PI oder TR gefärbt ist, auf das SIP und heben Sie den Tisch in Position. Beginnen Sie mit der Datenerfassung mit der niedrigsten Durchflussrate und beobachten Sie die Ereignisse in jedem Diagramm sorgfältig. Stellen Sie sicher, dass sich >90 % der Ereignisse in jedem Diagramm befinden und dass keine Ereignisse die VL2-H- und BL1-H-Kanäle sättigen.
- Fahren Sie mit dem Ausführen der Beispiele fort. Achten Sie darauf, mindestens 10.000 Ereignisse pro Probe aufzuzeichnen.
- Speichern Sie die Daten aus den Proben im .fcs-Dateiformat und exportieren Sie sie zur Analyse.
5. Datenanalyse der Ergebnisse der Durchflusszytometrie
HINWEIS: Dieser Datenanalyse-Workflow verwendet die FlowJo-Software. Wenn eine andere Durchflusszytometrie-Analysesoftware verwendet wird, befolgen Sie weiterhin die unten beschriebenen wichtigen Schritte mit softwaregeeigneten Tools. Um die Diagramme und das Gating zu visualisieren, siehe Ergänzende Abbildung S3 und Ergänzende Abbildung S4.
- Gate für lebende Zellen.
- Doppelklicken Sie auf das nicht gefärbte WT-Steuerelement, um ein Fenster für dieses Beispiel zu öffnen.
- Zeigen Sie die Daten als Histogramm entweder auf dem YL2-H-Kanal (bei Verwendung von PI) oder RL2-H (bei Verwendung von TR) an. Wechseln Sie zwischen diesem und Proben, die mit dem Viabilitätsfarbstoff gefärbt wurden, um die lebenden und toten Populationen zu identifizieren.
HINWEIS: Alle Ereignisse sollten nicht gefärbt werden, da dieser Probe kein Viabilitätsfarbstoff zugesetzt wurde. - Schaffung eines Winkelhalbierentors, das die lebende und die tote Bevölkerung trennt; nenne das linke Tor Live und das rechte Tor Dead. Wenden Sie dieses Gate auf alle Samples an und wechseln Sie dann zwischen den Samples, um sicherzustellen, dass dieses Gate für alle Samples richtig gezeichnet wird. Stellen Sie das Tor nach Bedarf ein.
- Tor für die Zellen.
- Doppelklicken Sie auf dem nicht markierten WT-Steuerelement auf das Live-Gate , um Ereignisse in diesem Gate anzuzeigen. Ändern Sie die x-Achse des Punktdiagramms in FSC-A und die y-Achse in SSC-A.
- Verwenden Sie das Werkzeug Polygon-Gate , um ein Gate um die Zellenpopulation zu zeichnen und es Zellen zu nennen. Achten Sie darauf, Trümmer/sterbende Zellen (in der Regel ganz links und unten im Punktdiagramm) und Aggregate (ganz rechts und oben im Punktdiagramm) auszuschließen.
- Wenden Sie dieses Gate unter dem Live-Gate für alle Proben an. Wechseln Sie zwischen den Proben, um sicherzustellen, dass das Gate die wahrscheinliche Zellpopulation für alle Proben umfasst, und nehmen Sie die erforderlichen Anpassungen vor. Achten Sie darauf, das Tor nach der Änderung erneut auf alle Proben anzuwenden.
HINWEIS: Die Verteilung der Zellpopulation ändert sich merklich zwischen ausgehungerten und nicht ausgehungerten Bedingungen bei FSC vs. SSC; Stellen Sie sicher, dass das Cell-Gate die Zellpopulation unter allen Bedingungen umfasst.
- Gate für Einzelzellen zur Erhöhung der Qualität von pH-Messungen.
- Doppelklicken Sie auf dem nicht gefärbten WT-Steuerelement auf das Cell-Gate , um Ereignisse in diesem Gate anzuzeigen. Ändern Sie die x-Achse des Punktdiagramms in FSC-A und die y-Achse in FSC-H.
- Suchen Sie in diesem Punktdiagramm nach einer diagonalen Verteilung einzelner Zellen mit Dubletten, die eine sekundäre Population unten rechts in der Singulett-Population bilden (siehe Ergänzende Abbildung S1 , drittes Diagramm). Zeichnen Sie mit dem Polygon-Gate-Werkzeug ein Gate um die Singulett-Ereignisse mit Ausnahme der Dublett-Population. Nennen Sie dieses Tor Singlets.
- Wenden Sie das Singulett-Gate unter dem Cell-Gate für alle Proben an. Wechseln Sie erneut zwischen den Stichproben, um sicherzustellen, dass das Gate die Dublett-Population ordnungsgemäß ausschließt, während die Singulett-Population einbezogen wird. Passen Sie es nach Bedarf an.
- Gate für fluoreszierende pHL-Zellen.
- Doppelklicken Sie in der ungefärbten WT-Kontrollstichprobe auf das Singulett-Gate , um ein Punktdiagramm für diese Population zu öffnen. Ändern Sie die x-Achse in BL1-H und die y-Achse in VL2-H.
- Der pH-Sensor pHluorin2 ist sowohl in VL2 als auch in BL1 fluoreszierend. Zeichnen Sie mit dem Werkzeug Polygontor ein diagonales Tor, das sich nach oben und rechts von der autofluoreszierenden Population unten links im Punktdiagramm erstreckt. Nennen Sie dieses Gatter pHL+.
- Wenden Sie das pHL+ -Gate für alle Proben unter dem Singulett-Gate an. Wechseln Sie zu einer pHL-Probe und passen Sie das Gate so an, dass Ereignisse mit höherer Fluoreszenzintensität sowohl in VL2-H als auch in BL1-H als in der WT-Steuerung enthalten sind. Stellen Sie sicher, dass dieses Gate diese fluoreszierende Population für alle Proben umfasst, da sich die Position dieser Population ändert, wenn sich der glykosomale pH-Wert ändert.
HINWEIS: Diese kleine, aber sichtbare Verschiebung in der pHL+-Population ist auf pH-abhängige Änderungen im Anregungsspektrum des Fluorophors zurückzuführen.
- Exportieren Sie die Statistiken.
- Klicken Sie auf Tabelleneditor | Bearbeitungsleiste | Spalte hinzufügen , um neue Statistiken für den Export hinzuzufügen.
HINWEIS: Wählen Sie für jede zu exportierende Statistik die entsprechende Statistik und die Population aus, aus der sie exportiert werden soll. Stellen Sie sicher, dass Sie den entsprechenden Parameter für anwendbare Statistiken auswählen, z. B. Median. Lassen Sie die Probe unverändert. - Fügen Sie Spalten mit den folgenden Statistiken hinzu: Gesamtanzahl (nicht belegt), pHL+-Anzahl, Live-Frequenz der Gesamtsumme (Prozentsatz basierend auf der Gesamtzahl der Ereignisse), pHL+ Frequenz des übergeordneten Elements (Prozentsatz basierend auf dem übergeordneten Gate), pHL+ Median VL2-H und pHL+ Median BL1-H.
- Klicken Sie auf den Tabelleneditor und ändern Sie die folgenden Exporteinstellungen: In Datei anzeigen und Text in CSV und wählen Sie dann das Dateiziel und den Namen. Klicken Sie auf Tabelle erstellen.
- Klicken Sie auf Tabelleneditor | Bearbeitungsleiste | Spalte hinzufügen , um neue Statistiken für den Export hinzuzufügen.
- Berechnen Sie das Fluoreszenzverhältnis.
- Speichern Sie die exportierte .csv-Datei als Tabellenkalkulationsdatei.
- Führen Sie eine Qualitätskontrollanalyse durch, indem Sie Folgendes über alle Stichproben im Experiment hinweg vergleichen: Anzahl der Ereignisse pro Stichprobe, Prozentsatz der Liveereignisse und Prozentsatz der pHL+-Ereignisse. Vergleichen Sie diese je nach Experiment visuell in Balken- oder Streudiagrammen.
- Beschriften Sie eine neue Spalte als pHL+ Median VL2/BL1. Teilen Sie für jede Probe den Medianwert VL2-H durch den Medianwert BL1-H, wie in Gleichung (1) gezeigt.
 (1)
(1)
- Verwenden Sie eine statistische Analysesoftware, um statistische Analysen anhand des Fluoreszenzverhältnisses durchzuführen.
6. pH-Biosensor-Kalibrierung
HINWEIS: Um gemessene Fluoreszenzverhältnisse in pH-Einheiten umzurechnen, kalibrieren Sie pHL-exprimierende Zellen mit Nigericin und Valinomycin. Nigericin ist ein K+/H+-Antiporter, ein Ionophor, das den pH-Wert über Membranen hinweg ausgleichen kann, wenn genügend K+ im Puffervorhanden ist 15. Nigericin wurde häufig zur Kalibrierung von pHluorin und anderen pH-Sensoren verwendet16,17. Da das peroxisomal lokalisierte pHluorin zuvor mit 10 μM Nigericin18 kalibriert wurde, entschieden wir uns für die Behandlung mit dieser Konzentration. Valinomycin ist ein Kaliumionophor und wurde (bei 4 μM) verwendet, um den pH-Wert über Mitochondrienmembranen auszugleichen19. Wir verwendeten 10 μM Valinomycin, um die pH-Gleichgewichtsaktivität von Nigericin zu unterstützen, indem wir sicherstellten, dass K+-Ionen über die Membranen hinweg ausgeglichen wurden. Während wir eine Nigericin-Valinomycin-Kombination verwendeten, kann Nigericin ausreichen, um den organellären pH-Wert auszugleichen.
- Bereiten Sie acht Lösungen von universellem Kalibrierpuffer (UCB; 15 mM MES, 15 mM HEPES und 130 mM KCl) mit jeweils einem anderen pH-Wert von 5 bis 8,5 vor.
- Zentrifugieren (RT, 10 min, 800-1.000 × g) acht separate Röhrchen mit 2 ml pHL-exprimierender BSF-Kultur (jeweils ~4 × 106 Zellen).
- Entfernen Sie den Überstand und resuspendieren Sie dann jedes Zellpellet in UCB bei unterschiedlichen pH-Werten.
- Führen Sie Nigericin und Valinomycin jeweils auf 10 μM ein. PI-Spitze auf 1 μg/ml.
- Inkubieren Sie die Zellen in jeder Lösung 15 Minuten lang.
- Führen Sie jede Probe auf einem Durchflusszytometer durch, um das Fluoreszenzverhältnis wie in den Schritten 4-5 beschrieben zu messen.
- Wiederholen Sie dieses Experiment noch zweimal, um drei biologische Replikate für jeden pH-Wert zu erhalten. Exportieren Sie Daten im .fcs-Format.
- Analysieren Sie die FCS-Dateien wie in den Schritten 5.1 bis 5.8 beschrieben. Verwenden Sie das gemessene Fluoreszenzverhältnis für jeden pH-Wert, um den glykosomalen pH-Wert in zukünftigen Experimenten mit Gleichung (2) zu interpolieren.
 (2)
(2)
HINWEIS: Ergänzende Abbildung S3 zeigt die Punktdiagramme und das Gating-Schema. Die Ergebnisse sind in der ergänzenden Tabelle S2 zu finden. Im Folgenden wird beschrieben, wie Sie den pH-Wert aus dem Fluoreszenzverhältnis mit GraphPad Prism interpolieren. Wenn Sie eine andere Statistiksoftware verwenden, befolgen Sie die gleichen wichtigen Schritte.- Öffnen Sie GraphPad Prism und erstellen Sie eine XY-Tabelle mit drei Y-Replikaten. Fügen Sie die pH-Werte in die x-Säule und die zugehörigen Fluoreszenzverhältniswerte in die y-replizierten Spalten ein.
- Klicken Sie auf das Diagramm, das der Tabelle zugeordnet ist. Wenn Sie das Diagramm anzeigen, klicken Sie auf Analysieren unter dem Menüband Analyse | Interpolieren Sie eine Standardkurve unter XY-Analysen.
- Wählen Sie Sigmoidal, 4PL, X ist log (Konzentration), da die pH-Einheiten auf einer logarithmischen Skala liegen. Die Parameter "Oben" und "Unten" sind die geschätzten oberen und unteren Plateaus. Wählen Sie Keine besondere Behandlung von Ausreißern.
HINWEIS: Die Software versucht, die Daten an das in Gleichung (2) und Schritt 6.9.3 beschriebene Modell anzupassen. Suchen Sie nach Hinweisen auf mangelnde Anpassung in der Ergebnistabelle und der Kurve im Diagramm. - Um den pH-Wert aus den Fluoreszenzverhältnissen in anderen Experimenten zu interpolieren, gehen Sie zur Tabelle mit den pHL-Kalibrierungsdaten. Fügen Sie die Fluoreszenzverhältniswerte unter den Kalibrierungsdaten in die y-Säule(n) ein. Geben Sie jedem y-Wert einen Titel, aber lassen Sie den x-Wert (den pH-Wert) leer, da er unbekannt ist.
- Klicken Sie in der Ergebnisgalerie auf die Interpolationsanalyse , und wechseln Sie dann zur Registerkarte Interpolierte X-Replikate . Suchen Sie nach den interpolierten pH-Werten, die neben den eingegebenen Fluoreszenzverhältniswerten aufgeführt werden.
HINWEIS: Die Software verwendet das Modell und die am besten angepassten Parameterwerte, die aus den Kalibrierungsdaten ermittelt wurden, um den pH-Wert aus den Fluoreszenzverhältnissen für Experimente mit unbekanntem pH-Wert zu interpolieren.
7. Glukosemangel und Addback-Zeitverläufe
- Induzieren Sie über Nacht 15 ml BSF pHL-Parasiten mit 1 μg/ml Doxycyclin, das bei 37 °C inkubiert wurde, wie in Schritt 1.1 beschrieben.
- Waschen Sie 15 ml induzierte pHL-Kultur in PBS, ergänzt mit 10 mM Glukose. Wiederholen Sie diesen Schritt 3x.
- Waschen Sie gleichzeitig 3 ml WT-Kultur in PBS 3x, wie in Schritt 3.2 beschrieben.
- Nach dem ersten Waschen, wenn die pHL-Probe in 1 ml PBS resuspendiert wird, entfernen Sie ein 0,1-ml-Aliquot der pHL-Zellsuspension, um 10 mM Glukose als nicht ausgehungerte Kontrolle zu halten.
- Nach dem letzten Waschen resuspendieren Sie die pHL-Probe in PBS, ergänzt mit 1 μg/ml PI.
- Erfassung von Durchflusszytometriedaten
- Beginnen Sie mit der Überwachung des glykosomalen pH-Werts sowohl für die ausgehungerten als auch für die nicht verhungerten Proben, indem Sie jede Probe je nach Experiment und Probe alle 5, 10, 30 oder 90 Minuten auf einem Durchflusszytometer messen. Erfassen Sie Durchflusszytometriedaten wie in den Schritten 4.1 bis 4.3 beschrieben, und stellen Sie sicher, dass die Spannungen und Diagramme vorher vorbereitet sind.
- Führen Sie die ungefleckte WEA-Steuerung zuerst bei 0 min aus.
- Führen Sie die nicht ausgehungerte Kontrollprobe alle 90 Minuten auf dem Zytometer durch, beginnend bei 0 Minuten. Führen Sie für den Hungerzeitverlaufstest die ausgehungerte Probe alle 10 Minuten ab 0 Minuten durch. Für den Glukose-Addback-Assay führen Sie die ausgehungerte Probe bei 0, 5, 10, 20, 30, 60 und 90 Minuten durch; Wiederholen Sie dann den Vorgang nach der Einführung von Glukose.
- Inkubieren Sie die Proben bei Raumtemperatur während des 90-minütigen Hungerzeitkurses und des 180-minütigen Glukose-Addback-Zeitkurses.
- Analysieren Sie die FCS-Dateien wie in den Schritten 5.1 bis 5.8 beschrieben.
HINWEIS: Ergänzende Abbildung S4 zeigt, wie das Gating durchgeführt wird und wie die Punktdiagramme aussehen sollten. Die ergänzende Tabelle S3 und die ergänzende Tabelle S4 zeigen die Ergebnisse des Glukosemangelzeitverlaufstests bzw. des Glukose-Addbackzeitverlaufstests.
8. Optimierung des Assays für das Wirkstoffscreening
- Waschen Sie ungefähr 32 ml pHL-exprimierende BSF-Parasiten und 3 ml WT-Zellen, beide bei etwa 2 × 106 Zellen/ml, 2x in PBS, wie zuvor beschrieben.
HINWEIS: Pro Well müssen mehr als 10.000 Ereignisse aufgezeichnet werden, um die Variabilität der gemessenen Fluoreszenzverhältnisse zu minimieren und die Z-Faktor-Statistik genau zu messen. Die dafür erforderliche Mindestkultur beträgt ca. 26 ml, aber wir empfehlen 32 ml für eine einfache Handhabung.- Nach dem ersten Waschen wird die pHL-Probe gleichmäßig in zwei Mikrozentrifugenröhrchen aufgeteilt.
- Nach den Waschungen resuspendieren Sie eine pHL-Probe in 18 ml PBS mit 5 mM Glukose, 0,1 % DMSO und TR. Resuspendieren Sie die andere pHL-Probe in 18 ml PBS plus 0,1 % DMSO und TR.
HINWEIS: Das DMSO wird verwendet, um die Pufferzusammensetzung in einem Wirkstoffscreening nachzuahmen, da Verbindungen typischerweise in DMSO gelöst werden. - Übertragen Sie diese beiden Zelllösungen in ein 12-Well-Reservoir, 9 ml pro Well.
- Verwenden Sie einen Pipettierroboter, um die Zelllösungen in separate Hälften einer 384-Well-Platte zu pipettieren, 80 μl pro Well.
- Inkubieren Sie die Platte 1,5 h lang bei Raumtemperatur, schütteln Sie sie leicht und wickeln Sie sie in Aluminiumfolie, um die Fluorophore vor Licht zu schützen.
- Führen Sie die Platte mit einem Durchflusszytometer aus, das 384-Well-Platten verarbeiten kann.
HINWEIS: Der folgende Workflow ist an einen Attune NxT mit Cytkick Max Auto Sampler angepasst. Wenn ein anderes Durchflusszytometer verwendet wird, befolgen Sie die wichtigsten Schritte.- Führen Sie die Platte mit der schnellsten Durchflussrate (1.000 μl/min) aus und aktivieren Sie den Boost-Modus. Erfassen Sie 20 μl/Well. Fügen Sie einen Mischzyklus und einen Spülzyklus zwischen jeder Vertiefung hinzu.
- Erstellen Sie Diagramme wie in Schritt 4.1.3 beschrieben.
- Führen Sie eine ungefärbte WT-Röhrensteuerung aus und optimieren Sie die Spannungen wie in den Schritten 4.1.3 und 4.1.4 beschrieben. Führen Sie eine ausgehungerte pHL-Röhrenprobe durch und optimieren Sie die VL2- und BL1-Spannungen wie in Schritt 4.1 beschrieben.
- Beginnen Sie mit der Erfassung der Platte auf dem Durchflusszytometer. Führen Sie die Platte horizontal, damit es keinen signifikanten Unterschied zwischen der ausgehungerten und der nicht ausgehungerten Probenvertiefung gibt. Stellen Sie sicher, dass die Plattenerfassung in ~1,5 h abgeschlossen ist.
- Exportieren Sie die FCS-Dateien, und analysieren Sie die Daten, wie in den Schritten 5.1 bis 5.8 beschrieben. Ermitteln Sie den Durchschnitt (AVG) und die Standardabweichung (SD) der Fluoreszenzverhältnisse für die Proben, die entweder mit Glukose (Glc) oder ohne Glukose (Hunger) behandelt wurden.
HINWEIS: Die Daten aus unseren Z-Faktor-Experimenten finden Sie in der ergänzenden Tabelle S5. - Berechnen Sie die Z-Faktor20-Statistik mit Gleichung (3).
 (3)
(3)

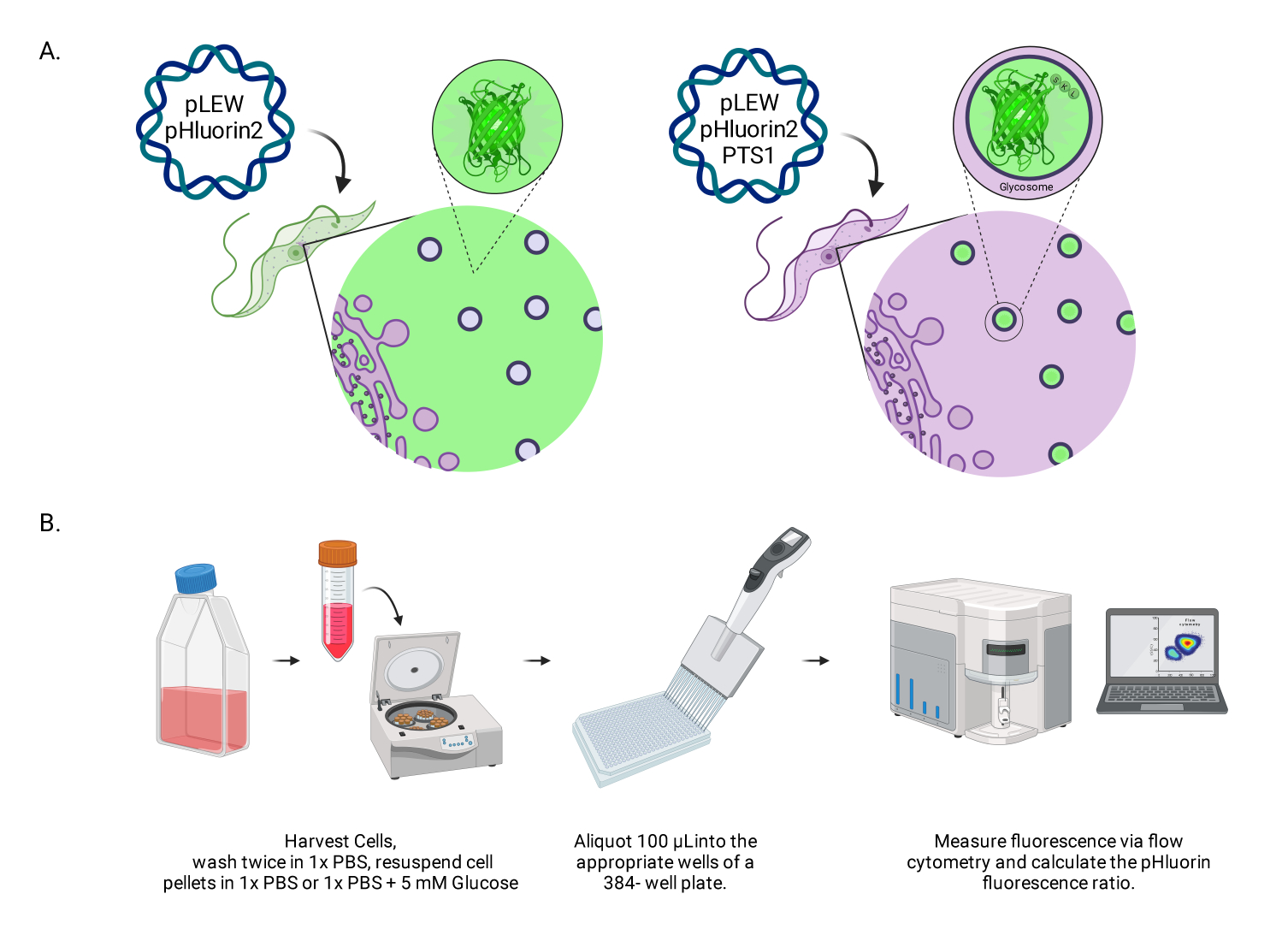
Abbildung 1: Diagramm der Methode zur Bewertung des glykosomalen pH-Werts in lebenden BSF-Trypanosomen. (A) Darstellung von Zelllinien, die den glykosomal lokalisierten pHluorin2-Sensor exprimieren. Der Einschluss einer peroxisomalen Targeting-Sequenz ermöglicht die Kontrolle über die Lokalisation. HINWEIS: Wir gehen davon aus, dass die Eliminierung des PTS-1 zu einer zytosolischen Lokalisation führen würde, die eine zukünftige Analyse des pH-Werts in diesem subzellulären Kompartiment ermöglicht. (B) Darstellung des Sensorvalidierungsassays. Abkürzung: BSF = Blutbahnform. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
HINWEIS: Die Z-Faktor-Statistik wird verwendet, um zu bestimmen, wie geeignet ein Assay für HTS ist. Werte zwischen 0,5 und 1,0 bedeuten im Allgemeinen, dass die Assay-Qualität für HTS akzeptabel ist.
Ergebnisse
pHLuorin2-PTS1-Lokalisierung an Glykosomen in BSF T. brucei
Um die subzelluläre Lokalisation des pHluorin2-PTS1 zu beurteilen, wurden die Parasiten Immunfluoreszenz-Assays unterzogen. Signal vom Transgen, das mit Antiseren kolokalisiert ist, die gegen ein Glykosomen-residentes Protein, Aldolase (TbAldolase), erhoben werden (Abbildung 2A). Der durchschnittliche Pearson-Korrelationskoeffizient der Kolokalisation zwischen Anti-TbAldolase und pHluorin2-PTS1 betrug 0...
Diskussion
Die Umweltwahrnehmung und die Reaktionsmechanismen im afrikanischen Trypanosom sind kaum verstanden. Es ist bekannt, dass Veränderungen der Nährstoffverfügbarkeit verschiedene Reaktionen im Parasiten auslösen, einschließlich der Ansäuerung von Glykosomen. Hier haben wir eine Methode zur Untersuchung der glykosomalen pH-Reaktion auf Umweltstörungen in lebenden Zellen unter Verwendung eines vererbbaren Proteinsensors, pHluorin2, und Durchflusszytometrie beschrieben.
Bei der Verwendung des...
Offenlegungen
Die Autoren erklären keine Interessenkonflikte.
Danksagungen
pHluorin2-PTS1 wurde von Twist Bioscience, die das Konstrukt in einem High-Copy-Klonierungsvektor zur Verfügung stellten, in pLEW100v5 kloniert; pLEW100v5 war ein Geschenk von Dr. George Cross. Antiserum gegen T. brucei Aldolase ist auf Anfrage bei Dr. Meredith T. Morris, Clemson University, erhältlich. Die Arbeit der JCM- und KAC-Laboratorien wurde teilweise durch eine Auszeichnung der National Institutes of Health (R01AI156382) unterstützt. SSP wurde von NIH 3R01AI156382 unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 50 mL Tissue Culture Flasks (Non-treated, sterile) | VWR | 10861-572 | |
| 75 cm2 Tissue Culture Flask (Non-Treated, sterile) | Corning | 431464U | |
| 80 µL flat-bottom 384-well plate | BrandTech | 781620 | |
| Amaxa Human T Cell Nucleofector Kit | Lonza | VPA-1002 | |
| Attune NxT Flow Cytometer | invitrogen by Thermo Fisher Scientific | A24858 | FlowJo software |
| BRANDplates 96-Well, flat bottom plate | Millipore Sigma | BR781662 | |
| Coloc 2 plugin of ImageJ | https://imagej.net/plugins/coloc-2 | ||
| CytKick Max Auto Sampler | invitrogen by Thermo Fisher Scientific | A42973 | |
| CytoFLEX Flow Cytometer | Beckman-Coulter | ||
| Electron Microscopy Sciences 16% Paraformaldehyde Aqueous Solution, EM Grade, 10 mL Ampoule | Fisher Scientific | 50-980-487 | |
| GraphPad Prism | statistical software | ||
| Nigericin (sodium salt) | Cayman Chemical | 11437 | |
| Nucleofector 2b | Lonza | Discontinued Product | |
| OP2 Liquid Handler | opentrons | OP2 | |
| poly-L-lysine, 0.1% (w/v) in H2O | Sigma Life Science | CAS:25988-63-0 | Pipetting robot for HTS assay |
| Thiazole Red (TO-PRO-3) | biotium | #40087 | We machined a custom acrylic plate stand so this brand of plate could be detected and used on our CytKick Max Auto Sampler |
| valinomycin | Cayman Chemical | 10009152 | Pipetting robot for HTS assay |
| For pH calibration | |||
| For pH calibration |
Referenzen
- Coley, A. F., Dodson, H. C., Morris, M. T., Morris, J. C. Glycolysis in the African trypanosome: Targeting enzymes and their subcellular compartments for therapeutic development. Molecular Biology International. 2011, 123702 (2011).
- Mcconville, M. J., Saunders, E. C., Kloehn, J., Dagley, M. J. Leishmania carbon metabolism in the macrophage phagolysosome- feast or famine. F1000Res. 4, 938 (2015).
- Parsons, M. Glycosomes: Parasites and the divergence of peroxisomal purpose. Molecular Microbiology. 53 (3), 717-724 (2004).
- Parsons, M., Furuya, T., Pal, S., Kessler, P. Biogenesis and function of peroxisomes and glycosomes. Molecular and Biochemical Parasitology. 115 (1), 19-28 (2001).
- Haanstra, J. R., Gonzalez-Marcano, E. B., Gualdron-Lopez, M., Michels, P. A. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochimica et Biophysica Acta. 1863 (5), 1038-1048 (2016).
- Allmann, S., Bringaud, F. Glycosomes: A comprehensive view of their metabolic roles in t. Brucei. International Journal of Biochemistry and Cell Biology. 85, 85-90 (2017).
- Dodson, H. C., Morris, M. T., Morris, J. C. Glycerol 3-phosphate alters Trypanosoma brucei hexokinase activity in response to environmental change. The Journal of Biological Chemistry. 286 (38), 33150-33157 (2011).
- Lin, S., Morris, M. T., Ackroyd, P. C., Morris, J. C., Christensen, K. A. Peptide targeted delivery of pH sensor for quantitative measurements of intraglycosomal pH in live Trypanosoma brucei. Biochemistry. 52 (21), 3629-3637 (2013).
- Mahon, M. J. Phluorin2: An enhanced, ratiometric, pH-sensitive green florescent protein. Advances in Bioscience and Biotechnology. 2 (3), 132-137 (2011).
- Wirtz, E., Leal, S., Ochatt, C., Cross, G. A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Molecular and Biochemical Parasitology. 99 (1), 89-101 (1999).
- . Restriction digest v.2 Available from: https://www.protocols.io/view/restriction-digest-nkqdg33pg25z/v2 (2018)
- . Ligation protocol with t4 DNA ligaase (m0202) v.3 Available from: https://www.protocols.io/view/ligation-protocol-with-t4-dna-ligase-m0202-95qpvorzv4o1/v3 (2021)
- Burkard, G., Fragoso, C. M., Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Molecular and Biochemical Parasitology. 153 (2), 220-223 (2007).
- Crowe, L. P., Wilkinson, C. L., Nicholson, K. R., Morris, M. T. Trypanosoma brucei pex13.2 is an accessory peroxin that functions in the import of peroxisome targeting sequence type 2 proteins and localizes to subdomains of the glycosome. mSphere. 5 (1), e00744 (2020).
- Kucejova, B., Kucej, M., Petrezselyova, S., Abelovska, L., Tomaska, L. A screen for nigericin-resistant yeast mutants revealed genes controlling mitochondrial volume and mitochondrial cation homeostasis. Genetics. 171 (2), 517-526 (2005).
- Huynh, M. H., Carruthers, V. B. Toxoplasma gondii excretion of glycolytic products is associated with acidification of the parasitophorous vacuole during parasite egress. PLoS Pathogens. 18 (5), e1010139 (2022).
- Lehoux, S., Abe, J., Florian, J. A., Berk, B. C. 14-3-3 binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. TheJournal of Biological Chemistry. 276 (19), 15794-15800 (2001).
- Jankowski, A., et al. In situ measurements of the ph of mammalian peroxisomes using the fluorescent protein phluorin. The Journal of Biological Chemistry. 276 (52), 48748-48753 (2001).
- Jankowski, A., Grinstein, S. A. A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. The Journal of Biological Chemistry. 274 (37), 26098-26104 (1999).
- Zhang, J. H., Chung, T. D., Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394 (6689), 192-195 (1998).
- Lin, S., et al. Ph regulation in glycosomes of procyclic form Trypanosoma brucei. The Journal of Biological Chemistry. 292 (19), 7795-7805 (2017).
- Ha, D. S., Schwarz, J. K., Turco, S. J., Beverley, S. M. Use of the green fluorescent protein as a marker in transfected Leishmania. Molecular and Biochemical Parasitology. 77 (1), 57-64 (1996).
- Kelly, J. M., Ward, H. M., Miles, M. A., Kendall, G. A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Research. 20 (15), 3963-3969 (1992).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten