
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Misurazione delle variazioni dinamiche del pH glisomiale nel Trypanosoma brucei vivente
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Descriviamo un metodo per studiare come il pH risponde ai segnali ambientali nei glicosomi della forma del flusso sanguigno dei tripanosomi africani. Questo approccio prevede l'utilizzo di un sensore di proteine ereditarie sensibile al pH in combinazione con la citometria a flusso per misurare la dinamica del pH, sia come saggio del corso temporale che in un formato di schermo ad alto rendimento.
Abstract
Il metabolismo del glucosio è fondamentale per il tripanosoma africano, Trypanosoma brucei, come processo metabolico essenziale e regolatore dello sviluppo del parassita. Poco si sa sulle risposte cellulari generate quando i livelli di glucosio ambientale cambiano. Sia nei parassiti del flusso sanguigno che in forma prociclica (stadio di insetto), i glicosomi ospitano la maggior parte della glicolisi. Questi organelli vengono rapidamente acidificati in risposta alla privazione di glucosio, che probabilmente si traduce nella regolazione allosterica degli enzimi glicolitici come l'esochinasi. Nel lavoro precedente, localizzare la sonda chimica utilizzata per effettuare misurazioni di pH era difficile, limitandone l'utilità in altre applicazioni.
Questo articolo descrive lo sviluppo e l'uso di parassiti che esprimono pHluorin2 glicosomialmente localizzato, un biosensore di pH proteico ereditabile. pHluorin2 è una variante raziometrica della pHluorin che mostra una diminuzione dell'eccitazione dipendente dal pH (acido) a 395 nm e contemporaneamente produce un aumento dell'eccitazione a 475 nm. I parassiti transgenici sono stati generati clonando il frame di lettura aperto di pHluorin2 nel vettore di espressione del tripanosoma pLEW100v5, consentendo l'espressione di proteine inducibili in entrambe le fasi del ciclo di vita. L'immunofluorescenza è stata utilizzata per confermare la localizzazione glicosale del biosensore pHluorin2, confrontando la localizzazione del biosensore con la proteina aldolasi glicosale residente. La risposta del sensore è stata calibrata a diversi livelli di pH incubando le cellule in una serie di tamponi che variavano in pH da 4 a 8, un approccio che abbiamo precedentemente utilizzato per calibrare un sensore di pH a base di fluoresceina. Abbiamo quindi misurato la fluorescenza di pHluorin2 a 405 nm e 488 nm utilizzando la citometria a flusso per determinare il pH glicosomiale. Abbiamo validato le prestazioni dei parassiti transgenici vivi che esprimono pHluorin2, monitorando il pH nel tempo in risposta alla deprivazione di glucosio, un noto fattore scatenante dell'acidificazione glicosale nei parassiti PF. Questo strumento ha una serie di potenziali applicazioni, tra cui il potenziale utilizzo nello screening dei farmaci ad alto rendimento. Oltre al pH glicosomiale, il sensore potrebbe essere adattato ad altri organelli o utilizzato in altri tripanosomatidi per comprendere la dinamica del pH nel contesto delle cellule vive.
Introduzione
I cinetoplastidi parassiti, come la maggior parte degli organismi viventi, si basano sul glucosio come componente fondamentale del metabolismo centrale del carbonio. Questo gruppo comprende organismi importanti dal punto di vista medico, come il tripanosoma africano, Trypanosoma brucei; il tripanosoma americano, T. cruzi; e parassiti del genere Leishmania. Il metabolismo del glucosio è fondamentale per la crescita del parassita nelle fasi del ciclo di vita dei patogeni. Ad esempio, quando viene privato del glucosio, la forma del flusso sanguigno (BSF) del tripanosoma africano muore rapidamente. In particolare, la glicolisi funge da unica fonte di ATP durante questa fase dell'infezione1. Anche i parassiti della Leishmania dipendono dal glucosio nell'ospite umano, con la fase del ciclo di vita dell'amastigote che risiede nei macrofagi dell'ospite che dipendono da questa fonte di carbonio per la crescita2.
Sebbene questi parassiti abbiano stili di vita distinti che coinvolgono diversi insetti vettori, condividono molti punti in comune nel modo in cui rispondono e consumano il glucosio. Ad esempio, questi parassiti localizzano la maggior parte degli enzimi glicolitici in perossisomi modificati chiamati glicosomi. Questo organello specifico per i cinetoplastidi è correlato ai perossisomi dei mammiferi sulla base di meccanismi biosintetici e morfologiaconservati 3,4,5,6.
La compartimentazione della maggior parte degli enzimi della via glicolitica nel glicosoma offre mezzi di regolazione della via specifici del parassita. Utilizzando una sonda chimica di pH, abbiamo stabilito che la privazione di nutrienti innesca una rapida acidificazione dei glicosomi del parassita in forma prociclica (PF) che si traduce in un'alterazione dell'attività enzimatica glicolitica attraverso l'esposizione di un sito di legame del regolatore allosterico sull'enzima glicolitico chiave esochinasi 7,8. Nel nostro lavoro precedente, la sonda chimica richiedeva una consegna costante per l'uso, limitandone l'utilità in altre applicazioni. Inoltre, le difficoltà a mantenere la distribuzione della sonda nel glicosoma nel BSF hanno limitato l'utilità dell'approccio per lo studio del pH glisomiale in quella fase di vita.
In questo studio, abbiamo utilizzato il biosensore proteico fluorescente pHluorin2 per monitorare la variazione del pH glicosomico in BSF T. brucei in risposta a segnali ambientali, tra cui la fame di glucosio9 (Figura 1). I risultati di questo lavoro suggeriscono che BSF T. brucei acidifica rapidamente i glicosomi in risposta alla fame in modo reversibile, in modo simile alle risposte che abbiamo osservato nei parassiti PF. Ci aspettiamo che questo biosensore migliorerà la nostra comprensione della regolazione glicolitica in T. brucei e nei parassiti correlati.
Access restricted. Please log in or start a trial to view this content.
Protocollo
L'utilizzo di t. brucei brucei 90-13 BSF tripanosomi, una linea di parassiti monomorfi, richiede considerazioni di sicurezza in quanto sono considerati organismi del gruppo di rischio 2 che dovrebbero essere maneggiati in strutture di livello di biosicurezza 2.
1. Coltura e trasfezione del tripanosoma
- Coltura di T. brucei brucei 90-13 tripanosomi BSF in terreno HMI-9 integrati con il 10% di FBS inattivato al calore e il 10% di Nu-Serum a 37 °C in 5% di CO210.
NOTA: Per mantenere la coltura sana, mantenere la densità cellulare tra 2 × 104 e 5 × 106 cellule/mL. - Clonazione di pHluorin2-PTS1 in pLEW100v5
- Sintetizzare commercialmente il frame di lettura aperto di pHluorin2 per produrre il gene con un'estensione di 3' che codifica per un linker a due glicine seguito dal tripeptide AKL, un tag di localizzazione PTS1.
- Clonare questo costrutto nel vettore di espressione del tripanosoma inducibile pLEW100v5 mediante digestione di restrizione. Digerire due volte sia il vettore di clonaggio contenente pHluorin2-PTS1 che pLEW100v5 utilizzando HindIII e BamHI11. Eseguire una fase di pulizia, preferibilmente mediante purificazione con gel di agarosio, per rimuovere gli enzimi di restrizione, la spina dorsale del vettore di clonaggio indesiderata e il frammento contenente il gene luciferasi asportato.
- Legare l'inserto cuscinetto pHluorin2 in pLEW100v5 digerito con T4 DNA Ligasi12. (vedere la figura supplementare S1 per uno schema per la clonazione).
- Sequenziare il plasmide mediante sequenziamento dell'intero plasmide di nuova generazione per verificare che l'inserto e il vettore siano legati correttamente e che non vengano introdotte mutazioni all'interno del gene pHluorin2-PTS1 durante il processo di clonaggio.
NOTA: La sequenza completa del plasmide è stata inviata a Addgene.org ed è stata assegnata #83680. - Linearizzare 20 μg del plasmide digerendo con 40 unità di NotI; quindi, trasfettare in parassiti BSF 90-13 tramite nucleofezione utilizzando il kit di cellule T umane di riferimento (vedi Tabella dei materiali). Selezionare per un'integrazione stabile come descritto da Burkard et al.13.
2. Colocalizzazione in immunofluorescenza di pHlourin2-PTS1
- Preparare i vetrini per microscopia trattandoli con poli-L-lisina 0,1% (p/v) in H2O per 10 min. Dopo aver rimosso la soluzione di poli-L-lisina, lavare il vetrino una volta con PBS.
- Per indurre l'espressione di pHluorin2-PTS1, trattare le cellule a 2 × 105 cellule/mL con tetraciclina o doxiciclina (1 μg/mL) 24 ore prima del prelievo. Pellet 2 × 106 parassiti parentali e indotti pLEWpHluorin2-PTS1 (pHL) mediante centrifugazione (temperatura ambiente [RT], 10 min, 1.000 × g) e lavare una volta con PBS.
- Risospendere le cellule in 200 μL di paraformaldeide al 2% appena preparata in PBS a base di una soluzione di grado EM al 16% fornita in commercio. Applicare le cellule del fissativo sul vetrino e lasciare riposare le cellule per 30 minuti. Lavare le cellule aderite 2 volte con una soluzione di lavaggio (siero di capra normale allo 0,1% in PBS).
- Applicare la soluzione di permeabilizzazione (0,5% Triton X-100 in PBS) sulle cellule e incubare per esattamente 30 minuti. Rimuovere la soluzione di permeabilizzazione e lavare una volta con abbondanti quantità di soluzione di lavaggio.
- Applicare la soluzione in blocco (10% di siero di capra normale e 0,1% di Triton X-100 in PBS) e incubare per 30 min.
- Diluire gli antisieri sollevati contro T. brucei aldolasi 1:500 in soluzione in blocco e applicarli sulle cellule14. Incubare per 1 ora a RT. Lavare i vetrini per 5 volte per 3-5 minuti min con soluzione di lavaggio.
- Diluire l'anticoniglio di capra Alexa fluor 568 1:1.000 in soluzione a blocchi e applicare sulle cellule. Incubare per 1 ora a RT. Lavare i vetrini per 5 volte per 3-5 minuti con la soluzione di lavaggio.
- Applicare il mezzo di montaggio e sigillare un vetrino coprioggetti sul vetrino.
- Fotografare le cellule con un obiettivo 100x (NA 1,4-0,7) e analizzare le immagini utilizzando ImageJ. Eseguire l'analisi di colocalizzazione di Pearson utilizzando il plug-in 'Coloc 2' per ImageJ. Vedere la Figura supplementare S2 per un campo di celle rappresentative. )
- Per completare questa operazione, aggiungi il file immagine a Figi e seleziona Immagini.
- Regolare la luminosità e il contrasto per ciascun canale in modo che non sia visibile lo sfondo.
- Modificare le immagini da 16 bit a 8 bit, unire le immagini e quindi ritagliarle in una singola cella con canali divisi.
- In Analizza e colocalizzazione, seleziona il plug-in Coloc2. Selezionare aldolasi (il canale rosso) come canale 1 e pHL (il canale verde) come canale 2 e fare clic su OK per avviare il calcolo della correlazione di Pearson.
3. Preparazione del campione per la citometria a flusso
- Indurre le cellule pHL con tetraciclina o doxiciclina (1 μg/mL) durante la notte.
- Pellet le cellule (~4 × 107 pHL e ~1 × 106 parentali) mediante centrifugazione (RT, 10 min, 1.000 × g). Rimuovere il surnatante e risospendere le cellule in 1 mL di PBS con o senza 10 mM di glucosio, a seconda che il campione sia affamato o meno. Per i saggi a tempo, risospendere in PBS integrato con 10 mM di glucosio per evitare che le cellule muoiano di fame fino al completamento dei lavaggi. Per il test di screening ad alto rendimento (HTS), risospendere in PBS senza glucosio per ridurre al minimo il carryover di glucosio; Ripetere i lavaggi altre due volte.
- Centrifugare le cellule per la quarta volta, rimuovere il surnatante e risospendere il pellet cellulare in PBS, PBS più 5 mM di glucosio o PBS più 10 mM di glucosio a seconda del trattamento. Integrare i campioni con 1 μg/mL di ioduro di propidio (PI) o 100 nM di rosso tiazolo (TR) per la determinazione di vivi/morti. Trasferire ogni campione in provette da 5 mL compatibili con il citometro a flusso.
4. Citometria a flusso
NOTA: Preparare l'esperimento su un citometro a flusso contenente i seguenti laser: 405 nm (viola), 488 nm (blu) e 561 nm (giallo) o 638 nm (rosso). Vedere la tabella supplementare S1 per i nomi comuni dei canali discussi di seguito.
- Per misurare la fluorescenza pHL, utilizzare i canali KO525 (VL2-H, eccitazione 405 nm, emissione 542/27 nm) e FITC (BL1-H, eccitazione 488 nm, emissione 530/30 nm). Per differenziare le cellule vive dalle cellule morte, utilizzare PI o TR; misura PI sul canale YL2-H (eccitazione 561 nm, emissione 620/25 nm). Misurare TR sul canale RL1-H (eccitazione a 638 nm, 660/10 BP).
- Per impostare l'esperimento sul software del citometro a flusso, creare i seguenti grafici: 1) istogramma del canale YL2-H (se si utilizza PI) o un istogramma del canale RL1-H (se si utilizza TR), 2) FSC-A vs SSC-A dot plot, 3) FSC-A vs FSC-H dot plot e 4) BL1-H vs VL2-H channel dot plot.
- Posizionare prima il controllo WT (parental cell-line) non colorato sulla porta di iniezione del campione (SIP) e sollevare il tavolino in posizione. Iniziare ad acquisire i dati sul citometro alla portata più bassa per dare il tempo di apportare le modifiche necessarie. Per evitare di segnare detriti spuri e cellule morte, iniziare a registrare gli eventi 5 secondi dopo l'inizio dell'acquisizione del campione.
- Regolare la tensione YL2 o RL1 in modo che il picco principale sia compreso tra 103-10 4 unità di intensità di fluorescenza relativa (RFI). Regolare le tensioni FSC e SSC in modo che il >90% degli eventi si adatti al dot plot. Regolare la soglia FSC per escludere la popolazione di detriti ma non le celle probabili.
- Regolare i canali VL2 e BL1 in modo che il picco primario sia compreso tra 103-10 4 unità RFI per il controllo WT non colorato.
- Posizionare il primo campione contenente pHL indotto colorato con PI o TR sul SIP e sollevare il tavolino in posizione. Iniziare ad acquisire i dati alla portata più bassa e osservare attentamente gli eventi in ogni grafico. Assicurarsi che il >90% degli eventi si trovi all'interno di ciascun grafico e che nessun evento stia saturando i canali VL2-H e BL1-H.
- Procedere con l'esecuzione degli esempi. Assicurarsi di registrare almeno 10.000 eventi per campione.
- Salvare i dati dei campioni in formato file .fcs ed esportarli per l'analisi.
5. Analisi dei dati dei risultati della citometria a flusso
NOTA: questo flusso di lavoro di analisi dei dati utilizza il software FlowJo. Se si utilizza un altro software di analisi della citometria a flusso, continuare a seguire i passaggi chiave descritti di seguito, utilizzando strumenti appropriati per il software. Per visualizzare i grafici e il gating, vedere la Figura supplementare S3 e la Figura supplementare S4.
- Aprire un nuovo layout e aprire i file .fcs acquisiti al punto 4.3. Trascinare e rilasciare i file .fcs nella finestra di layout.
- Cancello per cellule vive.
- Fare doppio clic sul controllo WT non colorato per aprire una finestra per l'esempio.
- Visualizzare i dati come istogramma sul canale YL2-H (se si utilizza PI) o RL2-H (se si utilizza TR). Alternare tra questo e i campioni colorati con il colorante di vitalità per identificare le popolazioni vive e morte.
NOTA: Tutti gli eventi non devono essere colorati poiché non è stato aggiunto un colorante di vitalità a questo campione. - Creare una porta a due settrici che divida le popolazioni vive e morte; denomina il cancello sinistro Vivo e il cancello destro Morto. Applicare questo cancello a tutti i campioni, quindi passare da un campione all'altro per assicurarsi che questo cancello sia disegnato in modo appropriato per tutti i campioni. Regolare il cancello secondo necessità.
- Cancello per le celle.
- Sul controllo WT non colorato, fare doppio clic sul cancello Live per visualizzare gli eventi in quel gate. Modificare l'asse x del dot plot in FSC-A e l'asse y in SSC-A.
- Usa lo strumento Cancello poligonale per disegnare un cancello attorno alla popolazione di celle e chiamarlo Celle. Fare attenzione a escludere i detriti/cellule morenti (in genere all'estrema sinistra e nella parte inferiore del diagramma a punti) e gli aggregati (all'estrema destra e in alto del grafico a punti).
- Applicare questo gate sotto il Live gate per tutti i campioni. Passare da un campione all'altro per assicurarsi che il gate comprenda la probabile popolazione cellulare per tutti i campioni e apportare le modifiche necessarie. Assicurarsi di riapplicare il gate a tutti i campioni dopo averlo modificato.
NOTA: La distribuzione della popolazione cellulare cambia notevolmente tra condizioni di fame e non di fame su FSC vs SSC; assicurarsi che il cancello cellulare comprenda la popolazione cellulare in tutte le condizioni.
- Gate per celle singole per aumentare la qualità delle misure di pH.
- Sul controllo WT non colorato, fare doppio clic sul gate Cell per visualizzare gli eventi in tale gate. Modificare l'asse x del grafico a punti in FSC-A e l'asse y in FSC-H.
- Cercare una distribuzione diagonale di singole cellule su questo grafico a punti con doppietti che formano una popolazione secondaria in basso a destra della popolazione di singoletto (vedere il terzo grafico della Figura S1 supplementare ). Utilizzando lo strumento Gate poligonale, disegna un gate attorno agli eventi singoletto escludendo la popolazione di doppietti. Assegna a questo cancello il nome Singlets.
- Applicare il cancello Singlets sotto il gate Cell per tutti i campioni. Anche in questo caso, passare da un campione all'altro per assicurarsi che il gate escluda correttamente la popolazione di doppietti includendo la popolazione di singoletto. Regolare secondo necessità.
- Gate per cellule pHL fluorescenti.
- Sul campione di controllo WT non colorato, fare doppio clic sul cancello Singlets per aprire un dot plot per quella popolazione. Modificare l'asse x in BL1-H e l'asse y in VL2-H.
- Il sensore di pH pHluorin2 è fluorescente sia in VL2 che in BL1. Usa lo strumento Gate poligonale per disegnare un gate di forma diagonale che si estende verso l'alto e verso destra lontano dalla popolazione autofluorescente in basso a sinistra del dot plot. Assegnare a questa porta il nome pHL+.
- Applicare il gate pHL+ sotto il gate Singlets per tutti i campioni. Passare a un campione pHL e regolare il gate per includere eventi con maggiore intensità di fluorescenza sia in VL2-H che in BL1-H rispetto al controllo WT. Assicurarsi che questo gate comprenda questa popolazione fluorescente per tutti i campioni, poiché la posizione di questa popolazione si sposterà al variare del pH glicosomiale.
NOTA: Questo piccolo ma visibile cambiamento nella popolazione pHL+ è dovuto a cambiamenti dipendenti dal pH nello spettro di eccitazione del fluoroforo.
- Esporta le statistiche.
- Fare clic su Editor tabelle | Barra di modifica | Aggiungi colonna per aggiungere nuove statistiche da esportare.
NOTA: per ogni statistica da esportare, scegliere la rispettiva statistica e la popolazione da cui esportarla. Assicurarsi di scegliere il parametro appropriato per le statistiche applicabili, ad esempio Mediana. Lasciare invariato il campione. - Aggiungi colonne con le seguenti statistiche: Conteggio totale (non valido), Conteggio pHL+, Frequenza in tempo reale del totale (percentuale basata sugli eventi totali), Frequenza pHL+ del genitore (percentuale basata sul gate padre), pHL+ Mediana VL2-H e Mediana pHL+ BL1-H.
- Fare clic sull'editor tabelle e modificare le seguenti impostazioni di esportazione: Visualizza su file e Testo su CSV, quindi scegliere la destinazione e il nome del file. Fare clic su Crea tabella.
- Fare clic su Editor tabelle | Barra di modifica | Aggiungi colonna per aggiungere nuove statistiche da esportare.
- Calcola il rapporto di fluorescenza.
- Salvare il file .csv esportato come foglio di calcolo.
- Eseguire l'analisi del controllo di qualità confrontando i seguenti elementi in tutti i campioni dell'esperimento: numero di eventi per campione, percentuale di eventi live e percentuale di eventi pHL+. Confrontali visivamente in grafici a barre o a dispersione a seconda dell'esperimento.
- Etichettare una nuova colonna come pHL+ Mediana VL2/BL1. Per ogni campione, dividere il valore mediano di VL2-H per il valore mediano di BL1-H come mostrato nell'equazione (1).
 (1)
(1)
- Utilizzare un software di analisi statistica per eseguire analisi statistiche utilizzando il rapporto di fluorescenza.
6. Calibrazione del biosensore di pH
NOTA: Per convertire i rapporti di fluorescenza misurati in unità di pH, calibrare le cellule che esprimono pHL utilizzando nigericina e valinomicina. La nigericina è un antiportatore K+/H+, uno ionoforo in grado di equilibrare il pH attraverso le membrane quando c'è abbastanza K+ nel tampone15. La nigericina è stata comunemente usata per calibrare la pHluorin e altri sensori di pH16,17. Poiché la pHluorin localizzata perossisomicamente era stata precedentemente calibrata utilizzando 10 μM di nigericina18, abbiamo scelto di trattare con quella concentrazione. La valinomicina è uno ionoforo di potassio ed è stata utilizzata (a 4 μM) per equilibrare il pH attraverso le membrane mitocondriali19. Abbiamo usato 10 μM di valinomicina per assistere l'attività di equilibrazione del pH della nigericina, assicurando che gli ioni K+ fossero equilibrati attraverso le membrane. Mentre abbiamo usato una combinazione di nigericina-valinomicina, la nigericina può essere sufficiente per equilibrare il pH organellare.
- Preparare otto soluzioni di tampone di calibrazione universale (UCB; 15 mM MES, 15 mM HEPES e 130 mM KCl) ciascuna a un pH diverso compreso tra 5 e 8,5.
- Centrifugare (RT, 10 min, 800-1.000 × g) otto provette separate da 2 mL di coltura BSF che esprime pHL (~4 × 106 cellule ciascuna).
- Rimuovere il surnatante e quindi risospendere ogni pellet cellulare in UCB a diversi valori di pH.
- Introdurre nigericina e valinomicina, ciascuna a 10 μM. Spike nel PI a 1 μg/mL.
- Incubare le cellule in ciascuna soluzione per 15 minuti.
- Eseguire ciascun campione su un citometro a flusso per misurare il rapporto di fluorescenza come descritto nei passaggi 4-5.
- Ripetere l'esperimento altre due volte per ottenere tre repliche biologiche per ogni valore di pH. Esporta i dati in formato .fcs.
- Analizzare i file con estensione fcs come descritto nei passaggi da 5.1 a 5.8. Utilizzare il rapporto di fluorescenza misurato per ciascun pH per interpolare il pH glicosomico in esperimenti futuri utilizzando l'equazione (2).
 (2)
(2)
NOTA: La Figura S3 supplementare mostra i diagrammi a punti e lo schema di gating. I risultati sono riportati nella Tabella supplementare S2. Di seguito viene descritto come interpolare il rapporto pH da fluorescenza utilizzando GraphPad Prism. Se si utilizza un altro software statistico, seguire gli stessi passaggi chiave.- Apri GraphPad Prism e crea una tabella XY con tre repliche y. Incollare i valori di pH nella colonna x e i valori del rapporto di fluorescenza associati nelle colonne a replica y.
- Fare clic sul grafico associato alla tabella. Quando si visualizza il grafico, fare clic su Analizza nella barra multifunzione Analisi | Interpolare una curva standard nelle analisi XY.
- Scegli Sigmoidal, 4PL, X è log (concentrazione) poiché le unità di pH sono su scala logaritmica. I parametri Top e Bottom sono i plateau superiori e inferiori stimati. Scegliere Nessuna gestione speciale dei valori anomali.
NOTA: Il software cercherà di adattare i dati al modello descritto nell'equazione (2) e nel passaggio 6.9.3. Cerca le prove della mancanza di adattamento nella tabella dei risultati e nella curva sul grafico. - Per interpolare il pH dai rapporti di fluorescenza in altri esperimenti, vai alla tabella con i dati di calibrazione del pHL. Incollare i valori del rapporto di fluorescenza sotto i dati di calibrazione nelle colonne y. Assegna un titolo a ciascun valore y, ma lascia vuoto il valore x (il pH) poiché è sconosciuto.
- Nella raccolta Risultati, fare clic sull'analisi di interpolazione , quindi passare alla scheda Repliche X interpolate . Cerca i valori di pH interpolati che verranno elencati insieme ai valori del rapporto di fluorescenza inseriti.
NOTA: Il software utilizza il modello e i valori dei parametri più adatti trovati dai dati di calibrazione per interpolare i rapporti di pH da fluorescenza per esperimenti in cui il pH è sconosciuto.
7. Digiuno di glucosio e corsi di tempo di addback
- Indurre 15 mL di parassiti BSF pHL per una notte con 1 μg/mL di doxiciclina incubata a 37 °C come descritto al punto 1.1.
- Lavare 15 mL di coltura pHL indotta in PBS integrata con 10 mM di glucosio. Ripetere questo passaggio 3 volte.
- Contemporaneamente, lavare 3 mL di coltura WT in PBS 3x come descritto al punto 3.2.
- Dopo il primo lavaggio, quando il campione di pHL viene risospeso in 1 mL di PBS, rimuovere un'aliquota di 0,1 mL di sospensione di cellule pHL per mantenere 10 mM di glucosio come controllo non affamato.
- Dopo il lavaggio finale, risospendere il campione di pHL in PBS integrato con 1 μg/mL PI.
- Acquisizione dei dati della citometria a flusso
- Iniziare a monitorare il pH glicosale sia per i campioni affamati che per quelli non affamati misurando ogni campione su un citometro a flusso ogni 5, 10, 30 o 90 minuti, a seconda dell'esperimento e del campione. Acquisire i dati della citometria a flusso come descritto nei passaggi da 4.1 a 4.3, assicurandosi che le tensioni e i grafici siano preparati in anticipo.
- Eseguire prima il controllo WT non colorato a 0 min.
- Eseguire il campione di controllo non affamato sul citometro ogni 90 minuti a partire da 0 minuti. Per il saggio del corso a tempo di fame, eseguire il campione affamato ogni 10 minuti a partire da 0 minuti. Per il test di addback del glucosio, eseguire il campione affamato a 0, 5, 10, 20, 30, 60 e 90 min; Quindi, ripetere dopo aver introdotto il glucosio.
- Incubare i campioni a temperatura ambiente durante il corso di 90 minuti di inedia e il corso di 180 minuti di glucosio Addback.
- Analizzare i file con estensione fcs come descritto nei passaggi da 5.1 a 5.8.
NOTA: la Figura S4 supplementare mostra come eseguire il gating e come dovrebbero apparire i grafici a punti. La Tabella supplementare S3 e la Tabella supplementare S4 mostrano rispettivamente i risultati del test del corso del tempo di carenza di glucosio e del test del corso del tempo di aggiunta del glucosio.
8. Ottimizzazione del test per lo screening dei farmaci
- Lavare circa 32 mL di parassiti BSF che esprimono pHL e 3 mL di cellule WT, entrambi a circa 2 × 106 cellule/mL, 2x in PBS come descritto in precedenza.
NOTA: È necessario registrare più di 10.000 eventi per pozzetto per ridurre al minimo la variabilità dei rapporti di fluorescenza misurati e misurare con precisione la statistica del fattore Z. La coltura minima necessaria per ottenere questo risultato è di circa 26 mL, ma si consiglia 32 mL per facilitare la manipolazione.- Dopo il primo lavaggio, separare equamente il campione di pHL in due provette per microcentrifuga.
- Dopo i lavaggi, risospendere un campione di pHL in 18 mL di PBS contenente 5 mM di glucosio, 0,1% di DMSO e TR. Risospendere l'altro campione di pHL in 18 mL di PBS più 0,1% di DMSO e TR.
NOTA: IL DMSO viene utilizzato per imitare la composizione del tampone in uno screening farmacologico poiché i composti sono tipicamente disciolti nel DMSO. - Trasferire queste due soluzioni cellulari in un serbatoio da 12 pozzetti, 9 mL per pozzetto.
- Utilizzare un robot di pipettaggio per pipettare le soluzioni cellulari in metà separate di una piastra da 384 pozzetti, 80 μL per pozzetto.
- Incubare la piastra a temperatura ambiente per 1,5 h, agitando delicatamente e avvolta in un foglio di alluminio per proteggere i fluorofori dalla luce.
- Far funzionare la piastra su un citometro a flusso in grado di eseguire piastre a 384 pozzetti.
NOTA: Il seguente flusso di lavoro è adattato a un Attune NxT con Cytkick Max Auto Sampler. Se si utilizza un altro citometro a flusso, continuare a seguire i passaggi chiave.- Per la piastra, funzionare alla portata più veloce (1.000 uL/min) e abilitare la modalità boost. Acquisire 20 μL/pozzetto. Includere un ciclo di miscelazione e un ciclo di risciacquo tra ogni pozzetto.
- Creare i grafici come descritto al punto 4.1.3.
- Eseguire un controllo del tubo WT non colorato e ottimizzare le tensioni come descritto nei passaggi 4.1.3 e 4.1.4. Eseguire un campione di tubo pHL affamato e ottimizzare le tensioni VL2 e BL1 come descritto al punto 4.1.
- Iniziare ad acquisire la piastra sul citometro a flusso. Far scorrere la piastra orizzontalmente in modo che non vi sia una differenza significativa di tempo di acquisizione tra i pozzetti per campioni affamati e non affamati. Assicurarsi che l'acquisizione della piastra sia terminata in ~1,5 h.
- Esportare i file .fcs e analizzare i dati come descritto nei passaggi da 5.1 a 5.8. Trova la media (AVG) e la deviazione standard (SD) dei rapporti di fluorescenza per i campioni trattati con glucosio (Glc) o senza glucosio (Starved).
NOTA: I dati dei nostri esperimenti con fattore Z sono disponibili nella Tabella supplementare S5. - Calcola la statistica del fattoreZ 20 usando l'equazione (3).
 (3)
(3)

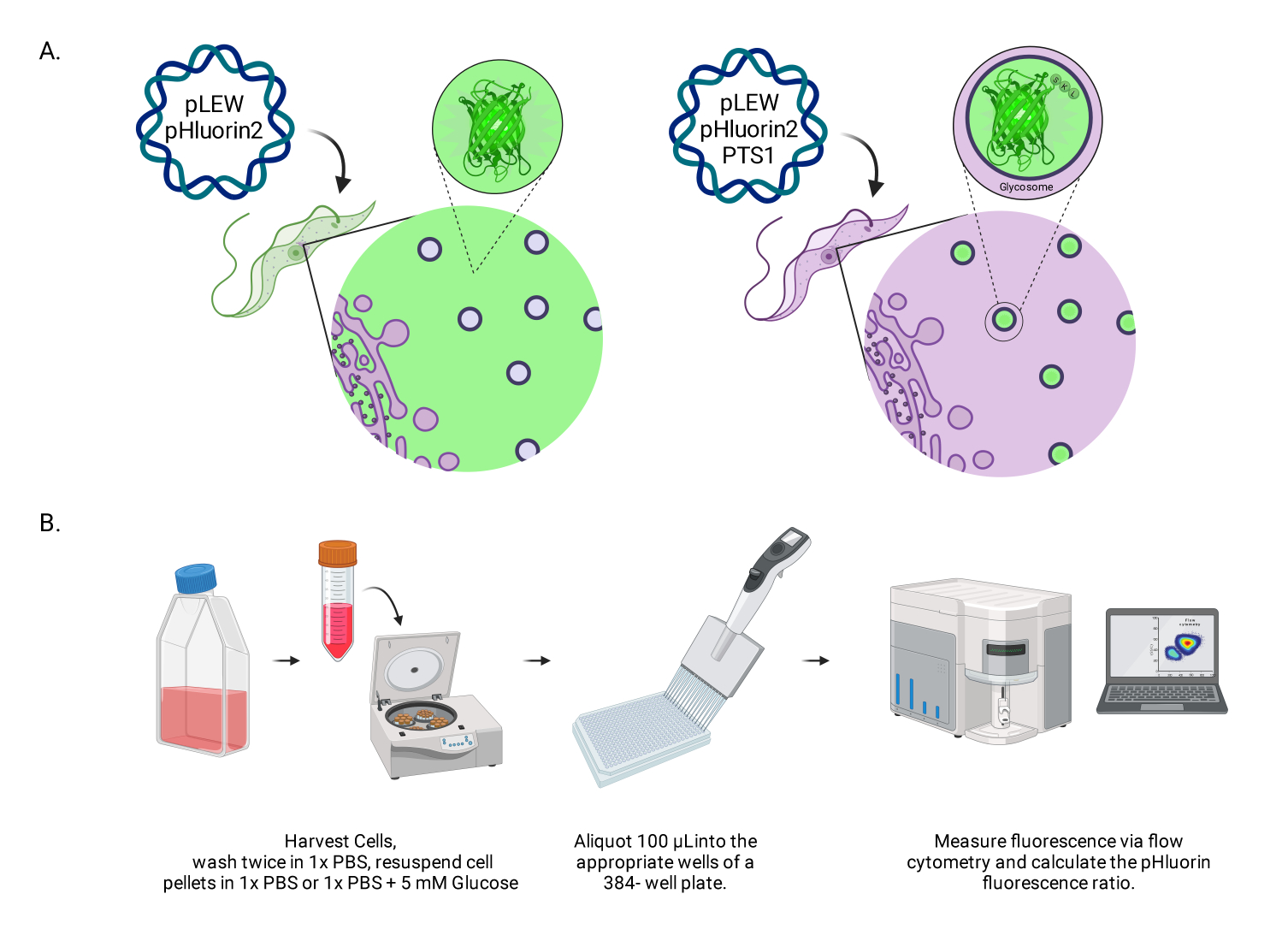
Figura 1: Diagramma del metodo per la valutazione del pH glicosomico nei tripanosomi vivi della BSF. (A) Rappresentazione di linee cellulari che esprimono il sensore di pHluorin2 localizzato glicosomialmente. L'inclusione di una sequenza di targeting perossisomiale fornisce il controllo sulla localizzazione. NOTA: Prevediamo che l'eliminazione del PTS-1 porterebbe alla localizzazione citosolica, consentendo future analisi del pH in quel compartimento subcellulare. (B) Rappresentazione del saggio di validazione del sensore. Abbreviazione: BSF = forma del flusso sanguigno. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
NOTA: La statistica del fattore Z viene utilizzata per determinare l'idoneità di un test per HTS. Valori compresi tra 0,5 e 1,0 indicano generalmente che la qualità del test è accettabile per l'HTS.
Access restricted. Please log in or start a trial to view this content.
Risultati
Localizzazione di pHLuorin2-PTS1 nei glicosomi in BSF T. brucei
Per valutare la localizzazione subcellulare della pHluorin2-PTS1, i parassiti sono stati sottoposti a saggi di immunofluorescenza. Segnale dal transgene colocalizzato con anti-sieri sollevato contro una proteina residente nel glicosoma, l'aldolasi (TbAldolasi) (Figura 2A). Il coefficiente medio di correlazione di Pearson di colocalizzazione tra anti-TbAldolasi e pHluorin2-PTS1 era 0,895, indicando ch...
Access restricted. Please log in or start a trial to view this content.
Discussione
La percezione ambientale e i meccanismi di risposta nel tripanosoma africano sono poco compresi. È noto che i cambiamenti nella disponibilità di nutrienti innescano diverse risposte nel parassita, inclusa l'acidificazione dei glicosomi. Qui, abbiamo descritto un metodo per studiare la risposta del pH glicosomiale alle perturbazioni ambientali nelle cellule viventi utilizzando un sensore di proteine ereditabili, pHluorin2 e citometria a flusso.
Ci sono diversi passaggi critici nell'uso del se...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori dichiarano di non avere conflitti di interesse.
Riconoscimenti
pHluorin2-PTS1 è stato clonato in pLEW100v5 da Twist Bioscience che ha fornito il costrutto in un vettore di clonazione ad alta copia; pLEW100v5 è stato un regalo del Dr. George Cross. L'antisiero contro l'aldolasi di T. brucei è disponibile presso la Dott.ssa Meredith T. Morris, Clemson University, su richiesta. Il lavoro dei laboratori JCM e KAC è stato parzialmente supportato da un premio del National Institutes of Health (R01AI156382). SSP è stato supportato da NIH 3R01AI156382.
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| 50 mL Tissue Culture Flasks (Non-treated, sterile) | VWR | 10861-572 | |
| 75 cm2 Tissue Culture Flask (Non-Treated, sterile) | Corning | 431464U | |
| 80 µL flat-bottom 384-well plate | BrandTech | 781620 | |
| Amaxa Human T Cell Nucleofector Kit | Lonza | VPA-1002 | |
| Attune NxT Flow Cytometer | invitrogen by Thermo Fisher Scientific | A24858 | FlowJo software |
| BRANDplates 96-Well, flat bottom plate | Millipore Sigma | BR781662 | |
| Coloc 2 plugin of ImageJ | https://imagej.net/plugins/coloc-2 | ||
| CytKick Max Auto Sampler | invitrogen by Thermo Fisher Scientific | A42973 | |
| CytoFLEX Flow Cytometer | Beckman-Coulter | ||
| Electron Microscopy Sciences 16% Paraformaldehyde Aqueous Solution, EM Grade, 10 mL Ampoule | Fisher Scientific | 50-980-487 | |
| GraphPad Prism | statistical software | ||
| Nigericin (sodium salt) | Cayman Chemical | 11437 | |
| Nucleofector 2b | Lonza | Discontinued Product | |
| OP2 Liquid Handler | opentrons | OP2 | |
| poly-L-lysine, 0.1% (w/v) in H2O | Sigma Life Science | CAS:25988-63-0 | Pipetting robot for HTS assay |
| Thiazole Red (TO-PRO-3) | biotium | #40087 | We machined a custom acrylic plate stand so this brand of plate could be detected and used on our CytKick Max Auto Sampler |
| valinomycin | Cayman Chemical | 10009152 | Pipetting robot for HTS assay |
| For pH calibration | |||
| For pH calibration |
Riferimenti
- Coley, A. F., Dodson, H. C., Morris, M. T., Morris, J. C. Glycolysis in the African trypanosome: Targeting enzymes and their subcellular compartments for therapeutic development. Molecular Biology International. 2011, 123702(2011).
- Mcconville, M. J., Saunders, E. C., Kloehn, J., Dagley, M. J. Leishmania carbon metabolism in the macrophage phagolysosome- feast or famine. F1000Res. 4, 938(2015).
- Parsons, M. Glycosomes: Parasites and the divergence of peroxisomal purpose. Molecular Microbiology. 53 (3), 717-724 (2004).
- Parsons, M., Furuya, T., Pal, S., Kessler, P. Biogenesis and function of peroxisomes and glycosomes. Molecular and Biochemical Parasitology. 115 (1), 19-28 (2001).
- Haanstra, J. R., Gonzalez-Marcano, E. B., Gualdron-Lopez, M., Michels, P. A. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochimica et Biophysica Acta. 1863 (5), 1038-1048 (2016).
- Allmann, S., Bringaud, F. Glycosomes: A comprehensive view of their metabolic roles in t. Brucei. International Journal of Biochemistry and Cell Biology. 85, 85-90 (2017).
- Dodson, H. C., Morris, M. T., Morris, J. C. Glycerol 3-phosphate alters Trypanosoma brucei hexokinase activity in response to environmental change. The Journal of Biological Chemistry. 286 (38), 33150-33157 (2011).
- Lin, S., Morris, M. T., Ackroyd, P. C., Morris, J. C., Christensen, K. A. Peptide targeted delivery of pH sensor for quantitative measurements of intraglycosomal pH in live Trypanosoma brucei. Biochemistry. 52 (21), 3629-3637 (2013).
- Mahon, M. J. Phluorin2: An enhanced, ratiometric, pH-sensitive green florescent protein. Advances in Bioscience and Biotechnology. 2 (3), 132-137 (2011).
- Wirtz, E., Leal, S., Ochatt, C., Cross, G. A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Molecular and Biochemical Parasitology. 99 (1), 89-101 (1999).
- Restriction digest v.2. , Available from: https://www.protocols.io/view/restriction-digest-nkqdg33pg25z/v2 (2018).
- Ligation protocol with t4 DNA ligaase (m0202) v.3. , Available from: https://www.protocols.io/view/ligation-protocol-with-t4-dna-ligase-m0202-95qpvorzv4o1/v3 (2021).
- Burkard, G., Fragoso, C. M., Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Molecular and Biochemical Parasitology. 153 (2), 220-223 (2007).
- Crowe, L. P., Wilkinson, C. L., Nicholson, K. R., Morris, M. T. Trypanosoma brucei pex13.2 is an accessory peroxin that functions in the import of peroxisome targeting sequence type 2 proteins and localizes to subdomains of the glycosome. mSphere. 5 (1), e00744(2020).
- Kucejova, B., Kucej, M., Petrezselyova, S., Abelovska, L., Tomaska, L. A screen for nigericin-resistant yeast mutants revealed genes controlling mitochondrial volume and mitochondrial cation homeostasis. Genetics. 171 (2), 517-526 (2005).
- Huynh, M. H., Carruthers, V. B. Toxoplasma gondii excretion of glycolytic products is associated with acidification of the parasitophorous vacuole during parasite egress. PLoS Pathogens. 18 (5), e1010139(2022).
- Lehoux, S., Abe, J., Florian, J. A., Berk, B. C. 14-3-3 binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. TheJournal of Biological Chemistry. 276 (19), 15794-15800 (2001).
- Jankowski, A., et al. In situ measurements of the ph of mammalian peroxisomes using the fluorescent protein phluorin. The Journal of Biological Chemistry. 276 (52), 48748-48753 (2001).
- Jankowski, A., Grinstein, S. A. A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. The Journal of Biological Chemistry. 274 (37), 26098-26104 (1999).
- Zhang, J. H., Chung, T. D., Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394 (6689), 192-195 (1998).
- Lin, S., et al. Ph regulation in glycosomes of procyclic form Trypanosoma brucei. The Journal of Biological Chemistry. 292 (19), 7795-7805 (2017).
- Ha, D. S., Schwarz, J. K., Turco, S. J., Beverley, S. M. Use of the green fluorescent protein as a marker in transfected Leishmania. Molecular and Biochemical Parasitology. 77 (1), 57-64 (1996).
- Kelly, J. M., Ward, H. M., Miles, M. A., Kendall, G. A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Research. 20 (15), 3963-3969 (1992).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati