
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Medición de los cambios dinámicos del pH glusómico en el Trypanosoma brucei vivo
* Estos autores han contribuido por igual
En este artículo
Resumen
Describimos un método para estudiar cómo responde el pH a las señales ambientales en los glicosomas del torrente sanguíneo de los tripanosomas africanos. Este enfoque implica un sensor de proteínas hereditarias sensible al pH en combinación con citometría de flujo para medir la dinámica del pH, tanto como un ensayo de curso temporal como en un formato de pantalla de alto rendimiento.
Resumen
El metabolismo de la glucosa es fundamental para el tripanosoma africano, Trypanosoma brucei, como un proceso metabólico esencial y regulador del desarrollo del parásito. Poco se sabe sobre las respuestas celulares que se generan cuando cambian los niveles ambientales de glucosa. Tanto en el torrente sanguíneo como en la forma procíclica (etapa de insecto), los glicosomas albergan la mayor parte de la glucólisis. Estos orgánulos se acidifican rápidamente en respuesta a la privación de glucosa, lo que probablemente resulta en la regulación alostérica de enzimas glucolíticas como la hexoquinasa. En trabajos anteriores, la localización de la sonda química utilizada para realizar mediciones de pH era un desafío, lo que limitaba su utilidad en otras aplicaciones.
En este trabajo se describe el desarrollo y uso de parásitos que expresan pHluorin2 localizada glucosómicamente, un biosensor de pH de proteína heredable. pHluorin2 es una variante radiométrica de pHluorin que muestra una disminución dependiente del pH (ácido) en la excitación a 395 nm mientras que simultáneamente produce un aumento en la excitación a 475 nm. Los parásitos transgénicos se generaron mediante la clonación del marco de lectura abierto de pHluorin2 en el vector de expresión de tripanosomas pLEW100v5, lo que permitió la expresión inducible de proteínas en cualquiera de las etapas del ciclo de vida. Se utilizó inmunofluorescencia para confirmar la localización glusómica del biosensor pHluorin2, comparando la localización del biosensor con la proteína residente glusómica aldolasa. La capacidad de respuesta del sensor se calibró a diferentes niveles de pH mediante la incubación de células en una serie de tampones que variaban en pH de 4 a 8, un enfoque que hemos utilizado previamente para calibrar un sensor de pH basado en fluoresceína. A continuación, medimos la fluorescencia de pHluorin2 a 405 nm y 488 nm mediante citometría de flujo para determinar el pH glusómico. Validamos el comportamiento de los parásitos transgénicos vivos que expresan pHluorin2, monitorizando el pH a lo largo del tiempo en respuesta a la privación de glucosa, un desencadenante conocido de la acidificación glusómica en parásitos PF. Esta herramienta tiene una serie de aplicaciones potenciales, incluida la posibilidad de ser utilizada en el cribado de drogas de alto rendimiento. Más allá del pH glusómico, el sensor podría adaptarse a otros orgánulos o usarse en otros tripanosomátidos para comprender la dinámica del pH en el entorno de células vivas.
Introducción
Los cinetoplástidos parásitos, como la mayoría de los organismos vivos, dependen de la glucosa como un componente fundamental del metabolismo central del carbono. Este grupo incluye organismos de importancia médica, como el tripanosoma africano, Trypanosoma brucei; el tripanosoma americano, T. cruzi; y parásitos del género Leishmania. El metabolismo de la glucosa es fundamental para el crecimiento del parásito en las etapas del ciclo de vida patógeno. Por ejemplo, cuando se le priva de glucosa, la forma del torrente sanguíneo (BSF) del tripanosoma africano muere rápidamente. En particular, la glucólisis sirve como la única fuente de ATP durante esta etapa de la infección1. Los parásitos de Leishmania también dependen de la glucosa en el huésped humano, y la etapa del ciclo de vida del amastigote que reside en los macrófagos del huésped depende de esta fuente de carbonopara su crecimiento.
Si bien estos parásitos tienen estilos de vida distintos que involucran diferentes insectos vectores, comparten muchos puntos en común en la forma en que responden y consumen glucosa. Por ejemplo, estos parásitos localizan la mayoría de las enzimas glucolíticas en peroxisomas modificados llamados glicosomas. Este orgánulo específico del cinetoplastido está relacionado con los peroxisomas de mamíferos en base a los mecanismos biosintéticos conservados y la morfología 3,4,5,6.
La compartimentación de la mayoría de las enzimas de la vía glucolítica en el glucosoma ofrece medios específicos para el parásito de regulación de la vía. Utilizando una sonda química de pH, establecimos que la privación de nutrientes desencadena una rápida acidificación de los glicosomas del parásito en forma procíclica (PF) que da lugar a una actividad enzimática glucolítica alterada a través de la exposición de un sitio de unión de un regulador alostérico en la enzima glucolítica clave hexoquinasa 7,8. En nuestro trabajo anterior, la sonda química requería una entrega constante para su uso, lo que limitaba su utilidad en otras aplicaciones. Además, los desafíos para mantener la distribución de la sonda en el glicosoma en el BSF limitaron la utilidad del enfoque para investigar el pH glusómico en esa etapa de la vida.
En este estudio, hemos utilizado el biosensor de proteína fluorescente pHluorin2 para monitorizar el cambio de pH glusómico en BSF T. brucei en respuesta a señales ambientales, incluida la inanición de glucosa9 (Figura 1). Los resultados de este trabajo sugieren que BSF T. brucei acidifica los glicosomas rápidamente en respuesta a la inanición de manera reversible, similar a las respuestas que hemos observado en los parásitos PF. Esperamos que este biosensor mejore nuestra comprensión de la regulación glucolítica en T. brucei y parásitos relacionados.
Protocolo
El uso de tripanosomas de T. brucei brucei 90-13 BSF, una línea de parásitos monomórficos, requiere tener en cuenta la seguridad, ya que se consideran organismos del Grupo de Riesgo 2 que deben manipularse en instalaciones de nivel de bioseguridad 2.
1. Cultivo y transfección de tripanosomas
- Cultivo de tripanosomas de T. brucei brucei 90-13 BSF en medio HMI-9 suplementado con FBS 10% inactivado por calor y 10% Nu-Serum a 37 °C en 5% CO210.
NOTA: Para mantener el cultivo sano, mantenga la densidad celular entre 2 × 104 y 5 × 106 células/mL. - Clonación de pHluorin2-PTS1 en pLEW100v5
- Sintetizar comercialmente el marco de lectura abierto de pHluorin2 para producir el gen con una extensión 3' que codifica un enlazador de dos glicinas seguido del tripéptido AKL, una etiqueta de localización de PTS1.
- Clone esta construcción en el vector de expresión de tripanosoma inducible pLEW100v5 mediante la síntesis de restricción. Digestión doble del vector de clonación que contiene pHluorin2-PTS1 y pLEW100v5 utilizando HindIII y BamHI11. Realice un paso de limpieza, preferiblemente mediante purificación en gel de agarosa, para eliminar las enzimas de restricción, la columna vertebral del vector de clonación no deseada y el fragmento que contiene el gen de la luciferasa extirpado.
- Ligue el inserto que contiene pHluorin2 en pLEW100v5 digerido con ADN ligasaT4 12. (véase la Figura Suplementaria S1 para ver un esquema de clonación).
- Secuenciar el plásmido mediante secuenciación de plásmido completo de próxima generación para verificar que el inserto y el vector estén ligados correctamente y que no se introduzcan mutaciones en el gen pHluorin2-PTS1 durante el proceso de clonación.
NOTA: La secuencia completa de plásmidos se ha enviado a Addgene.org y se le ha asignado #83680. - Linealizar 20 μg del plásmido digiriendo con 40 unidades de NotI; luego, transfectar en parásitos BSF 90-13 a través de nucleofección utilizando el kit de células T humanas referenciado (ver Tabla de Materiales). Seleccione para la integración estable como lo describen Burkard et al.13.
2. Colocalización por inmunofluorescencia de pHlourin2-PTS1
- Preparar portaobjetos de microscopía tratándolos con poli-L-lisina al 0,1% (p/v) enH2O durante 10 min. Después de retirar la solución de poli-L-lisina, lave el portaobjetos una vez con PBS.
- Para inducir la expresión de pHluorin2-PTS1, tratar las células a 2 × 105 células/ml con tetraciclina o doxiciclina (1 μg/ml) 24 h antes de la cosecha. Pellet 2 × 106 parásitos parentales e inducidos de pLEWpHluorin2-PTS1 (pHL) por centrifugación (temperatura ambiente [RT], 10 min, 1.000 × g) y lavar una vez con PBS.
- Resuspender las células en 200 μL de paraformaldehído al 2% recién preparado en PBS elaborado a partir de una solución de grado EM al 16% suministrada comercialmente. Aplique las células del fijador al portaobjetos y deje que las células se asienten durante 30 minutos. Lavar las células adheridas 2 veces con solución de lavado (0,1% de suero de cabra normal en PBS).
- Aplique la solución de permeabilización (Triton X-100 al 0,5% en PBS) a las células e incube durante exactamente 30 minutos. Retire la solución de permeabilización y lávese una vez con grandes cantidades de solución de lavado.
- Aplicar la solución en bloque (10% de suero normal de cabra y 0,1% de Triton X-100 en PBS) e incubar durante 30 min.
- Diluir los antisueros levantados contra T. brucei aldolasa 1:500 en solución de bloque y aplicarlos a las células14. Incubar durante 1 h a RT. Lave los portaobjetos 5 veces durante 3-5 minutos con solución de lavado.
- Diluir cabra anti-conejo Alexa fluor 568 1:1.000 en solución de bloque y aplicar sobre las células. Incubar durante 1 h a RT. Lave los portaobjetos 5 veces durante 3-5 minutos con solución de lavado.
- Aplique medio de montaje y selle un cubreobjetos a la guía.
- Visualice las celdas con un objetivo de 100x (NA 1.4-0.7) y analice las imágenes con ImageJ. Realice el análisis de colocalización de Pearson utilizando el plug-in 'Coloc 2' para ImageJ. Consulte la Figura complementaria S2 para ver un campo de células representativas. )
- Para completar esto, agregue el archivo de imagen a Fiji y seleccione Imágenes.
- Ajuste el brillo y el contraste de cada canal hasta un punto en el que no se vea el fondo.
- Cambie las imágenes de 16 bits a 8 bits, combine las imágenes y, a continuación, recórtelas en una sola celda con canales divididos.
- En Analizar y colocalización, seleccione el complemento Coloc2. Seleccione aldolasa (el canal rojo) como canal 1 y pHL (el canal verde) como canal 2 y haga clic en aceptar para iniciar el cálculo de la correlación de Pearson.
3. Preparación de la muestra para citometría de flujo
- Inducir células pHL con tetraciclina o doxiciclina (1 μg/mL) durante la noche.
- Granular las células (~4 × 107 pHL y ~1 × 106 parentales) por centrifugación (RT, 10 min, 1.000 × g). Retire el sobrenadante y vuelva a suspender las células en 1 mL de PBS, ya sea con o sin 10 mM de glucosa, dependiendo de si la muestra está hambrienta o no. Para los ensayos de curso temporal, vuelva a suspender en PBS suplementado con 10 mM de glucosa para evitar que las células mueran de hambre hasta que se completen los lavados. Para el ensayo de cribado de alto rendimiento (HTS), vuelva a suspender en PBS sin glucosa para minimizar el arrastre de glucosa; Repite los lavados dos veces más.
- Centrifugar las células por cuarta vez, eliminar el sobrenadante y volver a suspender el gránulo celular en PBS, PBS más 5 mM de glucosa o PBS más 10 mM de glucosa, según el tratamiento. Complementar las muestras con 1 μg/ml de yoduro de propidio (PI) o 100 nM de rojo tiazol (TR) para la determinación de vivos/muertos. Transfiera cada muestra a tubos de 5 ml compatibles con el citómetro de flujo.
4. Citometría de flujo
NOTA: Prepare el experimento en un citómetro de flujo que contenga los siguientes láseres: 405 nm (violeta), 488 nm (azul) y 561 nm (amarillo) o 638 nm (rojo). Consulte la Tabla complementaria S1 para conocer los nombres comunes de los canales que se analizan a continuación.
- Para medir la fluorescencia de pHL, utilice los canales KO525 (VL2-H, excitación 405 nm, emisión 542/27 nm) y FITC (BL1-H, excitación 488 nm, emisión 530/30 nm). Para diferenciar las células vivas de las células muertas, utilice PI o TR; medir PI en el canal YL2-H (excitación 561 nm, emisión 620/25 nm). Mida TR en el canal RL1-H (excitación de 638 nm, 660/10 BP).
- Para configurar el experimento en el software del citómetro de flujo, cree los siguientes gráficos: 1) histograma del canal YL2-H (si se utiliza PI) o un histograma del canal RL1-H (si se utiliza TR), 2) diagrama de puntos FSC-A frente a SSC-A, 3) diagrama de puntos FSC-A frente a FSC-H y 4) diagrama de puntos del canal BL1-H frente a VL2-H.
- Coloque primero el control WT (línea celular parental) sin teñir en el puerto de inyección de muestras (SIP) y levante la platina hasta su posición. Comience a adquirir datos en el citómetro con el caudal más bajo para dar tiempo a realizar los ajustes necesarios. Para evitar puntuar residuos espurios y células muertas, comience a registrar eventos 5 s después de que comience la adquisición de muestras.
- Ajuste el voltaje YL2 o RL1 para que el pico principal esté dentro de 103-10 4 unidades de intensidad de fluorescencia relativa (RFI). Ajuste los voltajes FSC y SSC para que el >90% de los eventos quepan en el diagrama de puntos. Ajuste el umbral FSC para excluir la población de escombros, pero no las células probables.
- Ajuste los canales VL2 y BL1 para que el pico primario esté dentro de 103-10 4 unidades RFI para el control WT sin teñir.
- Coloque la primera muestra que contenga pHL inducido teñida con PI o TR en el SIP y eleve la platina hasta su posición. Comience a adquirir datos con el caudal más bajo y observe cuidadosamente los eventos en cada gráfico. Asegúrese de que el >90% de los eventos estén dentro de cada gráfico y que ningún evento sature los canales VL2-H y BL1-H.
- Continúe con la ejecución de las muestras. Asegúrese de registrar al menos 10.000 eventos por muestra.
- Guarde los datos de las muestras en formato de archivo .fcs y expórtelos para su análisis.
5. Análisis de datos de los resultados de la citometría de flujo
NOTA: Este flujo de trabajo de análisis de datos utiliza el software FlowJo. Si se utiliza otro software de análisis de citometría de flujo, continúe siguiendo los pasos clave que se describen a continuación, utilizando las herramientas adecuadas para el software. Para visualizar las gráficas y las compuertas, consulte la Figura Suplementaria S3 y la Figura Suplementaria S4.
- Abra un nuevo diseño y abra los archivos .fcs adquiridos en el paso 4.3. Arrastre y suelte los archivos .fcs en la ventana de diseño.
- Puerta para células vivas.
- Haga doble clic en el control WT sin teñir para abrir una ventana para este ejemplo.
- Visualice los datos como un histograma en el canal YL2-H (si se utiliza PI) o en el canal RL2-H (si se utiliza TR). Alterna entre esto y las muestras teñidas con el tinte de viabilidad para identificar las poblaciones vivas y muertas.
NOTA: Todos los eventos deben estar sin teñir ya que no se agregó un tinte de viabilidad a esta muestra. - Crear una puerta bisectriz que divida las poblaciones vivas y muertas; Llama a la puerta de la izquierda Vive y a la puerta de la derecha Muerta. Aplique esta puerta a todas las muestras y, a continuación, alterne entre las muestras para asegurarse de que esta puerta se dibuja correctamente para todas las muestras. Ajuste la puerta según sea necesario.
- Puerta para las células.
- En el control WT sin teñir, haga doble clic en la puerta Live para ver los eventos en esa puerta. Cambie el eje x del diagrama de puntos a FSC-A y el eje y a SSC-A.
- Utilice la herramienta de puerta poligonal para dibujar una puerta alrededor de la población de celdas y asígnele el nombre Celdas. Tenga cuidado de excluir los escombros/células moribundas (normalmente en el extremo izquierdo y en la parte inferior del diagrama de puntos) y los agregados (en el extremo derecho y en la parte superior del diagrama de puntos).
- Aplique esta puerta debajo de la puerta Live para todas las muestras. Alterne entre muestras para asegurarse de que la puerta abarque la población celular probable para todas las muestras y realice los ajustes necesarios. Asegúrese de volver a aplicar la compuerta a todas las muestras después de modificarla.
NOTA: La distribución de la población celular cambia notablemente entre las condiciones de hambre y no inanición en FSC vs SSC; asegúrese de que la puerta de la celda abarque la población de celdas en todas las condiciones.
- Puerta para células individuales para aumentar la calidad de las mediciones de pH.
- En el control WT sin teñir, haga doble clic en la puerta de celda para ver los eventos de esa puerta. Cambie el eje x del diagrama de puntos a FSC-A y el eje y a FSC-H.
- Busque una distribución diagonal de celdas individuales en este diagrama de puntos con dobletes que forman una población secundaria en la parte inferior derecha de la población de singletes (consulte el tercer diagrama de la Figura Suplementaria S1 ). Con la herramienta de puerta de polígono, dibuje una puerta alrededor de los eventos singlete, excluyendo la población de doblete. Nombra esta puerta Singlets.
- Aplique la compuerta Singlets debajo de la compuerta Cell para todas las muestras. De nuevo, alterne entre las muestras para asegurarse de que la puerta excluye correctamente la población de dobletes e incluye la población de singletes. Ajústelo según sea necesario.
- Puerta para células fluorescentes de pHL.
- En la muestra de control WT sin teñir, haga doble clic en la puerta Singlets para abrir un diagrama de puntos para esa población. Cambie el eje x a BL1-H y el eje y a VL2-H.
- El sensor de pH pHluorin2 es fluorescente tanto en VL2 como en BL1. Utilice la herramienta de puerta poligonal para dibujar una puerta de forma diagonal que se extienda hacia la parte superior y hacia la derecha de la población autofluorescente en la parte inferior izquierda del diagrama de puntos. Asigne a esta puerta el nombre pHL+.
- Aplique la compuerta pHL+ debajo de la compuerta Singlets para todas las muestras. Cambie a una muestra de pHL y ajuste la puerta para incluir eventos con mayor intensidad de fluorescencia tanto en VL2-H como en BL1-H que en el control WT. Asegúrese de que esta puerta abarque esta población fluorescente para todas las muestras, ya que la posición de esta población cambiará a medida que cambie el pH glusómico.
NOTA: Este pequeño pero visible cambio en la población de pHL+ se debe a cambios dependientes del pH en el espectro de excitación del fluoróforo.
- Exporte las estadísticas.
- Haga clic en Editor de tablas | Barra de edición | Agregar columna para agregar nuevas estadísticas para exportar.
NOTA: Para cada estadística que se va a exportar, elija la estadística respectiva y la población desde la que se va a exportar. Asegúrese de elegir el parámetro adecuado para las estadísticas aplicables, como la mediana. Deje la muestra sin cambios. - Agregue columnas con las siguientes estadísticas: Recuento total (sin confirmar), Recuento de pHL+, Frecuencia en vivo del total (porcentaje basado en el total de eventos), Frecuencia de pHL+ de padre (porcentaje basado en la puerta principal), Mediana de pHL+ VL2-H y mediana de pHL+ BL1-H.
- Haga clic en el Editor de tablas y cambie la siguiente configuración de exportación: Mostrar a archivo y Texto a CSV y, a continuación, elija el destino y el nombre del archivo. Haga clic en Crear tabla.
- Haga clic en Editor de tablas | Barra de edición | Agregar columna para agregar nuevas estadísticas para exportar.
- Calcula la relación de fluorescencia.
- Guarde el archivo .csv exportado como un archivo de hoja de cálculo.
- Realice un análisis de control de calidad comparando lo siguiente en todas las muestras del experimento: número de eventos por muestra, porcentaje de eventos en vivo y porcentaje de eventos pHL+. Compáralos visualmente en diagramas de barras o de dispersión, según el experimento.
- Etiquete una nueva columna como pHL+ Mediana VL2/BL1. Para cada muestra, divida el valor mediano de VL2-H por el valor mediano de BL1-H, como se muestra en la ecuación (1).
 (1)
(1)
- Utilice un software de análisis estadístico para realizar análisis estadísticos utilizando la relación de fluorescencia.
6. Calibración del biosensor de pH
NOTA: Para convertir las relaciones de fluorescencia medidas en unidades de pH, calibre las células que expresan pHL utilizando nigericina y valinomicina. La nigericina es un antiportador de K+/H+, un ionóforo que puede equilibrar el pH a través de las membranas cuando hay suficiente K+ en el tampón15. La nigericina se ha utilizado comúnmente para calibrar la pHluorina y otros sensores de pH16,17. Como la pHluorina localizada peroxisomalmente se calibró previamente con 10 μM de nigericina18, se optó por tratar con esa concentración. La valinomicina es un ionóforo de potasio y se ha utilizado (a 4 μM) para equilibrar el pH a través de las membranas mitocondriales19. Utilizamos 10 μM de valinomicina para ayudar a la actividad de equilibrio del pH de la nigericina asegurando que los iones K+ se equilibraran a través de las membranas. Si bien usamos una combinación de nigericina y valinomicina, la nigericina puede ser suficiente para equilibrar el pH organelar.
- Prepare ocho soluciones de tampón de calibración universal (UCB; 15 mM MES, 15 mM HEPES y 130 mM KCl) cada una a un pH diferente que oscila entre 5 y 8,5.
- Centrífuga (RT, 10 min, 800-1.000 × g) ocho tubos separados de 2 mL de cultivo de BSF que expresan pHL (~4 × 106 células cada uno).
- Retire el sobrenadante y luego vuelva a suspender cada gránulo celular en UCB a diferentes valores de pH.
- Introducir nigericina y valinomicina, cada una a 10 μM. Pico en PI a 1 μg/mL.
- Incubar las células en cada solución durante 15 min.
- Ejecute cada muestra en un citómetro de flujo para medir la relación de fluorescencia como se describe en los pasos 4-5.
- Repita este experimento dos veces más para obtener tres réplicas biológicas para cada valor de pH. Exporte datos en formato .fcs.
- Analice los archivos .fcs como se describe en los pasos 5.1 a 5.8. Utilice la relación de fluorescencia medida para cada pH para interpolar el pH glusómico en futuros experimentos utilizando la ecuación (2).
 (2)
(2)
NOTA: La Figura Suplementaria S3 muestra los diagramas de puntos y el esquema de compuertas. Los resultados se pueden encontrar en la Tabla Suplementaria S2. A continuación se describe cómo interpolar el pH a partir de la relación de fluorescencia utilizando GraphPad Prism. Si utiliza otro software estadístico, siga los mismos pasos clave.- Abra GraphPad Prism y cree una tabla XY con tres réplicas y. Pegue los valores de pH en la columna x y sus valores de relación de fluorescencia asociados en las columnas de replicación y.
- Haga clic en el gráfico asociado a la tabla. Al ver el gráfico, haga clic en Analizar en la cinta Análisis | Interpolar una curva estándar en análisis XY.
- Elija Sigmoidal, 4PL, X es log(concentración) ya que las unidades de pH están en una escala logarítmica. Los parámetros Superior e Inferior son las mesetas superior e inferior estimadas. Elija No hay manejo especial de valores atípicos.
NOTA: El software tratará de ajustar los datos al modelo descrito en la ecuación (2) y en el paso 6.9.3. Busque evidencia de falta de ajuste en la tabla de resultados y la curva en el gráfico. - Para interpolar el pH a partir de las proporciones de fluorescencia en otros experimentos, vaya a la tabla con los datos de calibración de pHL. Pegue los valores de la relación de fluorescencia debajo de los datos de calibración en la(s) columna(s) y. Asigne un título a cada valor y, pero deje el valor x (el pH) en blanco, ya que se desconoce.
- En la galería de resultados, haga clic en el análisis de interpolación y luego vaya a la pestaña Réplicas X interpoladas . Busque los valores de pH interpolados que se enumerarán junto con los valores de relación de fluorescencia introducidos.
NOTA: El software utiliza el modelo y los valores de los parámetros de mejor ajuste encontrados en los datos de calibración para interpolar el pH de las proporciones de fluorescencia para experimentos en los que se desconoce el pH.
7. Cursos de tiempo de inanición y adición de glucosa
- Inducir 15 mL de parásitos BSF pHL durante la noche con 1 μg/mL de doxiciclina incubada a 37 °C como se describe en el paso 1.1.
- Lavar 15 mL de cultivo de pHL inducido en PBS suplementado con 10 mM de glucosa. Repita este paso 3 veces.
- Al mismo tiempo, lavar 3 ml de cultivo de WT en PBS 3x como se describe en el paso 3.2.
- Después del primer lavado, cuando la muestra de pHL se resuspende en 1 mL de PBS, retire una alícuota de 0,1 mL de suspensión de células de pHL para mantener 10 mM de glucosa como un control sin inanición.
- Después del lavado final, vuelva a suspender la muestra de pHL en PBS suplementada con 1 μg/mL PI.
- Adquisición de datos de citometría de flujo
- Comience a monitorear el pH glusómico tanto para las muestras hambrientas como para las no hambrientas midiendo cada muestra en un citómetro de flujo cada 5, 10, 30 o 90 minutos, según el experimento y la muestra. Adquiera datos de citometría de flujo como se describe en los pasos 4.1 a 4.3, asegurándose de que los voltajes y los gráficos estén preparados de antemano.
- Ejecute primero el control WT sin manchar a los 0 min.
- Ejecute la muestra de control sin hambre en el citómetro cada 90 minutos a partir de los 0 minutos. Para el ensayo de curso temporal de inanición, ejecute la muestra sin presencia cada 10 minutos a partir de los 0 minutos. Para el ensayo de adición de glucosa, ejecute la muestra sin presencia a los 0, 5, 10, 20, 30, 60 y 90 minutos; Luego, repita después de introducir glucosa.
- Incubar las muestras a temperatura ambiente durante el curso de tiempo de inanición de 90 minutos y el curso de tiempo de adición de glucosa de 180 minutos.
- Analice los archivos .fcs como se describe en los pasos 5.1 a 5.8.
NOTA: La Figura Suplementaria S4 muestra cómo realizar la compuerta y cómo deben verse los diagramas de puntos. La Tabla Suplementaria S3 y la Tabla Suplementaria S4 muestran los resultados del ensayo de tiempo de inanición de glucosa y el ensayo de tiempo de adición de glucosa, respectivamente.
8. Optimización del ensayo para el cribado de fármacos
- Lave aproximadamente 32 mL de parásitos BSF que expresan pHL y 3 mL de células WT, ambos a aproximadamente 2 × 106 células/mL, 2 veces en PBS como se describió anteriormente.
NOTA: Es necesario registrar más de 10.000 eventos por pocillo para minimizar la variabilidad de las relaciones de fluorescencia medidas y medir con precisión el estadístico del factor Z. El cultivo mínimo necesario para lograr esto es de aproximadamente 26 mL, pero recomendamos 32 mL para facilitar la manipulación.- Después del primer lavado, separe la muestra de pHL en partes iguales en dos tubos de microcentrífuga.
- Después de los lavados, vuelva a suspender una muestra de pHL en 18 ml de PBS que contenga 5 mM de glucosa, 0,1 % de DMSO y TR. Vuelva a suspender la otra muestra de pHL en 18 ml de PBS más 0,1 % de DMSO y TR.
NOTA: El DMSO se utiliza para imitar la composición del tampón en una prueba de detección de fármacos, ya que los compuestos suelen disolverse en el DMSO. - Transfiera estas dos soluciones celulares a un depósito de 12 pocillos, 9 ml por pocillo.
- Utilice un robot de pipeteo para pipetear las soluciones celulares en mitades separadas de una placa de 384 pocillos, 80 μL por pocillo.
- Incubar la placa a temperatura ambiente durante 1,5 h, agitándola suavemente y envolviéndola en papel de aluminio para proteger los fluoróforos de la luz.
- Haga funcionar la placa en un citómetro de flujo capaz de ejecutar placas de 384 pocillos.
NOTA: El siguiente flujo de trabajo está adaptado a un Attune NxT con Cytkick Max Auto Sampler. Si se utiliza otro citómetro de flujo, continúe siguiendo los pasos clave.- Para la placa, funcione al caudal más rápido (1.000 uL/min) y habilite el modo boost. Adquiere 20 μL/pocillo. Incluya un ciclo de mezcla y un ciclo de enjuague entre cada pocillo.
- Cree gráficos como se describe en el paso 4.1.3.
- Ejecute un control de tubo WT sin manchar y optimice los voltajes como se describe en los pasos 4.1.3 y 4.1.4. Ejecute una muestra de tubo de pHL sin presencia y optimice los voltajes VL2 y BL1 como se describe en el paso 4.1.
- Comience a adquirir la placa en el citómetro de flujo. Haga funcionar la placa horizontalmente para que no haya una diferencia significativa en el tiempo de adquisición entre los pocillos de muestra hambrientos y no hambrientos. Asegúrese de que la adquisición de la placa finalice en ~1,5 h.
- Exporte los archivos .fcs y analice los datos como se describe en los pasos 5.1 a 5.8. Encuentre el promedio (AVG) y la desviación estándar (DE) de las proporciones de fluorescencia para las muestras tratadas con glucosa (Glc) o sin glucosa (Starved).
NOTA: Los datos de nuestros experimentos con el factor Z se pueden encontrar en la Tabla Suplementaria S5. - Calcule el estadístico del factorZ 20 usando la ecuación (3).
 (3)
(3)

Figura 1: Diagrama del método para puntuar el pH glusómico en tripanosomas vivos de BSF. (A) Representación de líneas celulares que expresan el sensor pHluorin2 localizado glusómicamente. La inclusión de una secuencia de diana peroxisomal proporciona control sobre la localización. NOTA: Anticipamos que la eliminación de PTS-1 conduciría a la localización citosólica, lo que permitiría un análisis futuro del pH en ese compartimento subcelular. (B) Representación del ensayo de validación del sensor. Abreviatura: BSF = forma del torrente sanguíneo. Haga clic aquí para ver una versión más grande de esta figura.
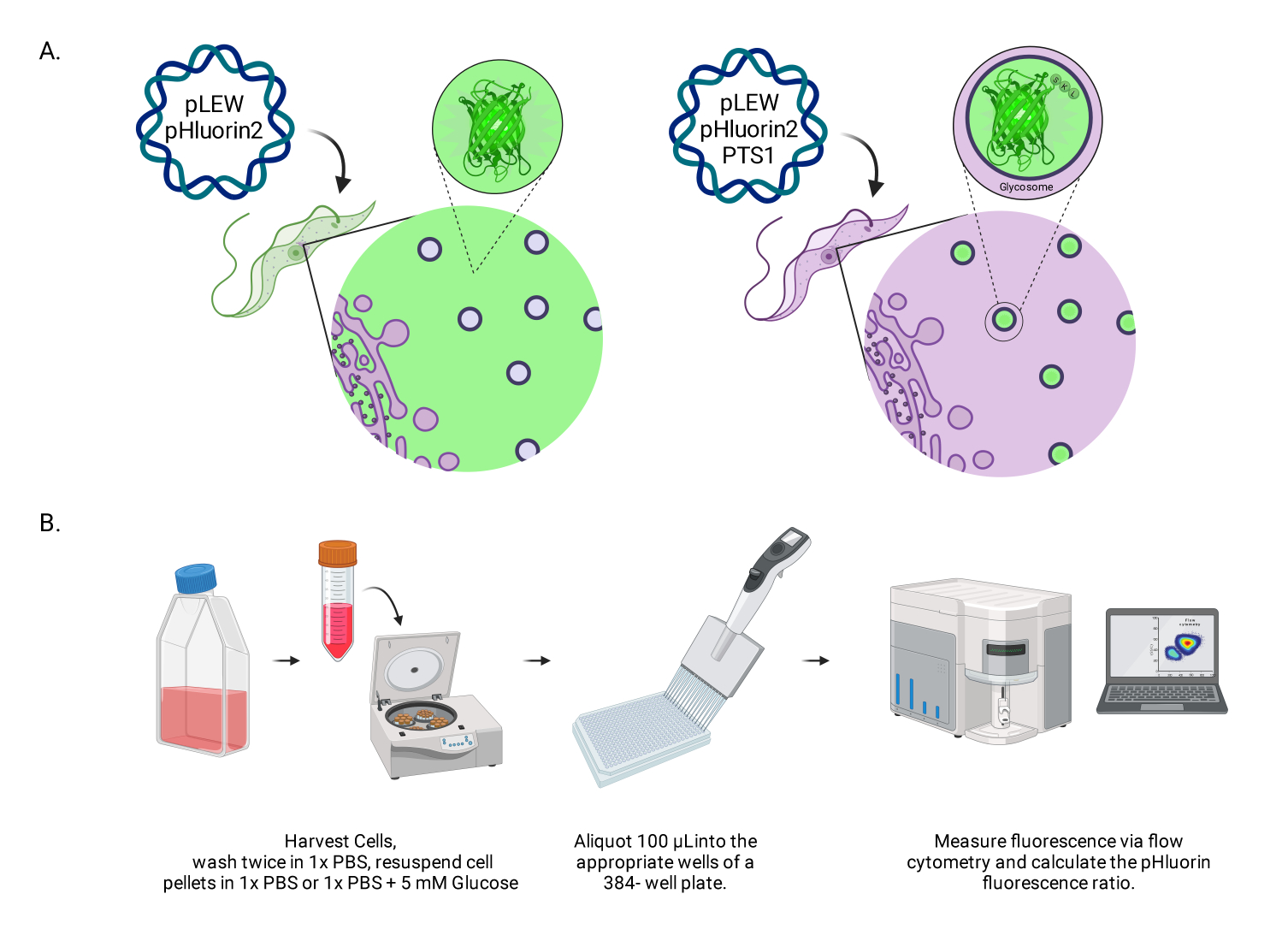{kind=link}
NOTA: El estadístico del factor Z se utiliza para determinar la idoneidad de un ensayo para HTS. Los valores entre 0,5 y 1,0 generalmente significan que la calidad del ensayo es aceptable para HTS.
Resultados
Localización de pHLuorin2-PTS1 en glicosomas en BSF T. brucei
Para evaluar la localización subcelular de la pHluorina2-PTS1, los parásitos se sometieron a ensayos de inmunofluorescencia. Señal del transgén colocalizada con antisueros frente a una proteína residente en el glicosoma, la aldolasa (TbAldolase) (Figura 2A). El coeficiente de correlación promedio de Pearson de colocalización entre la anti-TbAldolasa y la pHluorina2-PTS1 fue de 0,895, lo que ind...
Discusión
La percepción ambiental y los mecanismos de respuesta en el tripanosoma africano son poco conocidos. Se sabe que los cambios en la disponibilidad de nutrientes desencadenan diversas respuestas en el parásito, incluida la acidificación de los glicosomas. Aquí, hemos descrito un método para estudiar la respuesta del pH glusómico a las perturbaciones ambientales en células vivas utilizando un sensor de proteína heredable, pHluorin2, y citometría de flujo.
Hay varios pasos críticos en el...
Divulgaciones
Los autores declaran no tener conflictos de intereses.
Agradecimientos
pHluorin2-PTS1 fue clonado en pLEW100v5 por Twist Bioscience, que proporcionó la construcción en un vector de clonación de alta copia; pLEW100v5 fue un regalo del Dr. George Cross. El antisuero contra la aldolasa de T. brucei está disponible en la Dra. Meredith T. Morris, de la Universidad de Clemson, previa solicitud. El trabajo de los laboratorios JCM y KAC fue parcialmente apoyado por un premio de los Institutos Nacionales de Salud (R01AI156382). SSP fue apoyado por NIH 3R01AI156382.
Materiales
| Name | Company | Catalog Number | Comments |
| 50 mL Tissue Culture Flasks (Non-treated, sterile) | VWR | 10861-572 | |
| 75 cm2 Tissue Culture Flask (Non-Treated, sterile) | Corning | 431464U | |
| 80 µL flat-bottom 384-well plate | BrandTech | 781620 | |
| Amaxa Human T Cell Nucleofector Kit | Lonza | VPA-1002 | |
| Attune NxT Flow Cytometer | invitrogen by Thermo Fisher Scientific | A24858 | FlowJo software |
| BRANDplates 96-Well, flat bottom plate | Millipore Sigma | BR781662 | |
| Coloc 2 plugin of ImageJ | https://imagej.net/plugins/coloc-2 | ||
| CytKick Max Auto Sampler | invitrogen by Thermo Fisher Scientific | A42973 | |
| CytoFLEX Flow Cytometer | Beckman-Coulter | ||
| Electron Microscopy Sciences 16% Paraformaldehyde Aqueous Solution, EM Grade, 10 mL Ampoule | Fisher Scientific | 50-980-487 | |
| GraphPad Prism | statistical software | ||
| Nigericin (sodium salt) | Cayman Chemical | 11437 | |
| Nucleofector 2b | Lonza | Discontinued Product | |
| OP2 Liquid Handler | opentrons | OP2 | |
| poly-L-lysine, 0.1% (w/v) in H2O | Sigma Life Science | CAS:25988-63-0 | Pipetting robot for HTS assay |
| Thiazole Red (TO-PRO-3) | biotium | #40087 | We machined a custom acrylic plate stand so this brand of plate could be detected and used on our CytKick Max Auto Sampler |
| valinomycin | Cayman Chemical | 10009152 | Pipetting robot for HTS assay |
| For pH calibration | |||
| For pH calibration |
Referencias
- Coley, A. F., Dodson, H. C., Morris, M. T., Morris, J. C. Glycolysis in the African trypanosome: Targeting enzymes and their subcellular compartments for therapeutic development. Molecular Biology International. 2011, 123702 (2011).
- Mcconville, M. J., Saunders, E. C., Kloehn, J., Dagley, M. J. Leishmania carbon metabolism in the macrophage phagolysosome- feast or famine. F1000Res. 4, 938 (2015).
- Parsons, M. Glycosomes: Parasites and the divergence of peroxisomal purpose. Molecular Microbiology. 53 (3), 717-724 (2004).
- Parsons, M., Furuya, T., Pal, S., Kessler, P. Biogenesis and function of peroxisomes and glycosomes. Molecular and Biochemical Parasitology. 115 (1), 19-28 (2001).
- Haanstra, J. R., Gonzalez-Marcano, E. B., Gualdron-Lopez, M., Michels, P. A. Biogenesis, maintenance and dynamics of glycosomes in trypanosomatid parasites. Biochimica et Biophysica Acta. 1863 (5), 1038-1048 (2016).
- Allmann, S., Bringaud, F. Glycosomes: A comprehensive view of their metabolic roles in t. Brucei. International Journal of Biochemistry and Cell Biology. 85, 85-90 (2017).
- Dodson, H. C., Morris, M. T., Morris, J. C. Glycerol 3-phosphate alters Trypanosoma brucei hexokinase activity in response to environmental change. The Journal of Biological Chemistry. 286 (38), 33150-33157 (2011).
- Lin, S., Morris, M. T., Ackroyd, P. C., Morris, J. C., Christensen, K. A. Peptide targeted delivery of pH sensor for quantitative measurements of intraglycosomal pH in live Trypanosoma brucei. Biochemistry. 52 (21), 3629-3637 (2013).
- Mahon, M. J. Phluorin2: An enhanced, ratiometric, pH-sensitive green florescent protein. Advances in Bioscience and Biotechnology. 2 (3), 132-137 (2011).
- Wirtz, E., Leal, S., Ochatt, C., Cross, G. A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Molecular and Biochemical Parasitology. 99 (1), 89-101 (1999).
- . Restriction digest v.2 Available from: https://www.protocols.io/view/restriction-digest-nkqdg33pg25z/v2 (2018)
- . Ligation protocol with t4 DNA ligaase (m0202) v.3 Available from: https://www.protocols.io/view/ligation-protocol-with-t4-dna-ligase-m0202-95qpvorzv4o1/v3 (2021)
- Burkard, G., Fragoso, C. M., Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Molecular and Biochemical Parasitology. 153 (2), 220-223 (2007).
- Crowe, L. P., Wilkinson, C. L., Nicholson, K. R., Morris, M. T. Trypanosoma brucei pex13.2 is an accessory peroxin that functions in the import of peroxisome targeting sequence type 2 proteins and localizes to subdomains of the glycosome. mSphere. 5 (1), e00744 (2020).
- Kucejova, B., Kucej, M., Petrezselyova, S., Abelovska, L., Tomaska, L. A screen for nigericin-resistant yeast mutants revealed genes controlling mitochondrial volume and mitochondrial cation homeostasis. Genetics. 171 (2), 517-526 (2005).
- Huynh, M. H., Carruthers, V. B. Toxoplasma gondii excretion of glycolytic products is associated with acidification of the parasitophorous vacuole during parasite egress. PLoS Pathogens. 18 (5), e1010139 (2022).
- Lehoux, S., Abe, J., Florian, J. A., Berk, B. C. 14-3-3 binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. TheJournal of Biological Chemistry. 276 (19), 15794-15800 (2001).
- Jankowski, A., et al. In situ measurements of the ph of mammalian peroxisomes using the fluorescent protein phluorin. The Journal of Biological Chemistry. 276 (52), 48748-48753 (2001).
- Jankowski, A., Grinstein, S. A. A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. The Journal of Biological Chemistry. 274 (37), 26098-26104 (1999).
- Zhang, J. H., Chung, T. D., Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Miesenbock, G., De Angelis, D. A., Rothman, J. E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 394 (6689), 192-195 (1998).
- Lin, S., et al. Ph regulation in glycosomes of procyclic form Trypanosoma brucei. The Journal of Biological Chemistry. 292 (19), 7795-7805 (2017).
- Ha, D. S., Schwarz, J. K., Turco, S. J., Beverley, S. M. Use of the green fluorescent protein as a marker in transfected Leishmania. Molecular and Biochemical Parasitology. 77 (1), 57-64 (1996).
- Kelly, J. M., Ward, H. M., Miles, M. A., Kendall, G. A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Research. 20 (15), 3963-3969 (1992).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados