
Method Article
Identificación del receptor de superficie celular mediante pantallas genéticas CRISPR/Cas9 de escala de genoma
En este artículo
Resumen
Este manuscrito describe un enfoque de cribado basado en células a escala del genoma para identificar interacciones extracelulares entre receptores y ligand.
Resumen
La comunicación intercelular mediada por interacciones directas entre receptores de superficie celulares incrustados en membrana es crucial para el desarrollo normal y el funcionamiento de organismos multicelulares. Sin embargo, la detección de estas interacciones sigue siendo técnicamente difícil. Este manuscrito describe un enfoque sistemático de detección genética de knockout CRISPR/Cas9 a escala del genoma que revela las vías celulares necesarias para eventos específicos de reconocimiento de superficie celular. Este ensayo utiliza proteínas recombinantes producidas en un sistema de expresión de proteínas de mamíferos como ávidas sondas de unión para identificar socios de interacción en una pantalla genética basada en células. Este método se puede utilizar para identificar los genes necesarios para las interacciones de la superficie celular detectadas por sondas de unión recombinantes correspondientes a los ectodominios de los receptores incrustados en la membrana. Es importante destacar que, dada la naturaleza a escala del genoma de este enfoque, también tiene la ventaja de no sólo identificar el receptor directo, sino también los componentes celulares que se requieren para la presentación del receptor en la superficie celular, proporcionando así información valiosa sobre la biología del receptor.
Introducción
Las interacciones extracelulares por las proteínas receptoras de superficie celular dirigen procesos biológicos importantes como la organización del tejido, el reconocimiento del patógeno huésped y la regulación inmune. Investigar estas interacciones es de interés para la comunidad biomédica más amplia, porque los receptores de membrana son objetivos procesables de terapias entregadas sistemáticamente como anticuerpos monoclonales. A pesar de su importancia, el estudio de estas interacciones sigue siendo técnicamente difícil. Esto se debe principalmente a que los receptores incrustados en membrana son anfipáticos, lo que dificulta el aislamiento de las membranas biológicas para la manipulación bioquímica, y sus interacciones se caracterizan por las débiles afinidades de interacción(KDs en el rango de M-mM)1. En consecuencia, muchos métodos de uso común no son adecuados para detectar esta clase de interacciones proteicas1,2.
Una gama de métodos se ha desarrollado para investigar específicamente las interacciones extracelulares receptor-ligand que tienen en cuenta sus propiedades bioquímicas únicas3. Algunos de estos enfoques implican expresar todo el ectodominio de un receptor como proteína recombinante soluble en sistemas basados en mamíferos o células de insectos para asegurarse de que estas proteínas contienen modificaciones posttranslacionales que son estructuralmente importantes, como los glicanos y los enlaces disulfuros. Para superar el enlace de baja afinidad, los ectodominios a menudo se oligomerizan para aumentar su avidez de enlace. Los ectodominios proteicos ávidos se han utilizado con éxito como sondas de unión para identificar socios de interacción en pantallas de interacción proteína-proteína recombinante directa4,5,6,7. Si bien son ampliamente exitosos, los métodos basados en proteínas recombinantes requieren que el ectodominio de un receptor de membrana se produzca como una proteína soluble. Por lo tanto, sólo es generalmente aplicable a proteínas que contienen una región extracelular contigua (por ejemplo, de paso único tipo I, tipo II o anclado A GPI) y no es generalmente adecuado para complejos receptores y proteínas de membrana que abarcan la membrana varias veces.
Las técnicas de clonación de expresiones en las que una biblioteca de DNA complementarios (cDNAs) se transfecta en las células y se ha probado para un fenotipo de ganancia de unión también se han utilizado para identificar interacciones proteínas y proteínas extracelulares8. La disponibilidad de grandes colecciones de plásmidos de expresión de ADNc clonados y secuenciados en los últimos años ha facilitado métodos en los que las líneas celulares que sobreexpresan los cDNAs que codifican los receptores de superficie celular se examinan para la unión de proteínas recombinantes para identificar interacciones9,,10. Los enfoques basados en la sobreexpresión de ADNn. a diferencia de los métodos basados en proteínas recombinantes, ofrecen la posibilidad de identificar interacciones en el contexto de la membrana plasmática. Sin embargo, el éxito del uso de construcciones de expresión de ADNc depende de la capacidad de las células para sobreexpresar la proteína en la forma correctamente doblada, pero esto a menudo requiere factores accesorios celulares como transportadores, chaperones y el ensamblaje oligomérico correcto. Por lo tanto, la transfectación de un solo ADNc podría no ser suficiente para lograr la expresión de la superficie celular.
Las técnicas de cribado que utilizan construcciones de ADNc o sondas de proteína recombinante consumen muchos recursos y requieren grandes colecciones de ADNb o bibliotecas de proteínas recombinantes. Recientemente se han utilizado métodos basados en espectrometría de masas diseñados específicamente para identificar interacciones extracelulares que no requieren el montaje de bibliotecas grandes. Sin embargo, estas técnicas requieren manipulación química de la superficie celular, que puede alterar la naturaleza bioquímica de las moléculas presentes en la superficie de las células y actualmente sólo son aplicables para interacciones mediadas por proteínas glicosiladas11,12. La mayoría de los métodos actualmente disponibles también se centran en gran medida en las interacciones entre proteínas, ignorando en gran medida la contribución del microambiente de membrana, incluyendo moléculas como glicanos, lípidos y colesterol.
El reciente desarrollo de la focalización bialélica altamente eficiente utilizando enfoques basados en CRISPR ha permitido bibliotecas a escala del genoma de células que carecen de genes definidos en un solo grupo que se pueden examinar de manera sistemática e imparcial para identificar los componentes celulares implicados en diferentes contextos, incluyendo la disección de procesos de señalización celular, la identificación de perturbaciones que confieren resistencia a fármacos, toxinas y patógenos, y la determinación de la especificidad de los anticuerpos13,14,15,16. Aquí, describimos un ensayo de cribado de células eliminatorias basado en CRISPR a escala genómico que proporciona una alternativa a los enfoques bioquímicos actuales para identificar interacciones extracelulares entre receptores y ligand. Este enfoque de identificación de interacciones mediadas por receptores de membrana por pantallas genéticas es particularmente adecuado para los investigadores que tienen un interés centrado en los ligandos individuales porque evita la necesidad de compilar grandes bibliotecas de cDNAs o proteínas recombinantes.
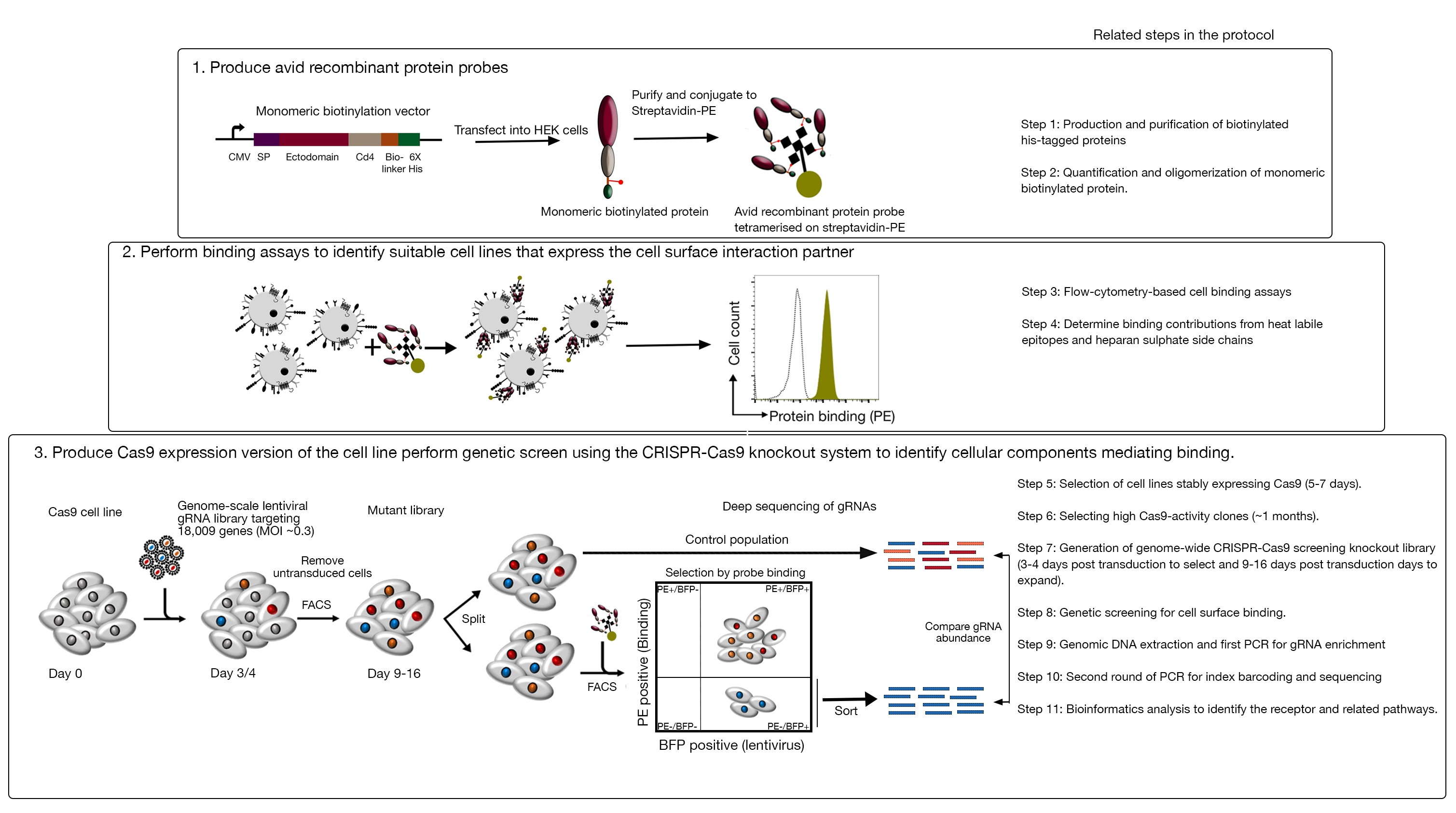
Este ensayo consta de tres pasos principales: 1) Las sondas de unión a proteínas recombinantes altamente ávidas que consisten en las regiones extracelulares de un receptor de interés se producen y se utilizan en ensayos de unión basados en fluorescencia de flujo basados en fluorescencia; 2) Los ensayos de unión se utilizan para identificar una línea celular que expresa el socio de interacción de la sonda de proteína recombinante; 3) Se produce una versión que expresa Cas9 de la línea celular que interactúa con la proteína de interés y se realiza una pantalla eliminatoria basada en CRISPR/Cas9 a escala del genoma (Figura 1). En esta pantalla genética, la unión de una proteína recombinante a las líneas celulares se utiliza como un fenotipo medible en el que las células dentro de la biblioteca knockout que han perdido la capacidad de unir la sonda se ordenan mediante la clasificación celular activada basada en fluorescencia (FACS) y los genes que causaron la pérdida del fenotipo de unión identificado por la secuenciación. En principio, se identifican los genes que codifican el receptor responsable de la unión de la sonda ávida y los necesarios para su visualización de la superficie celular.
El primer paso de este protocolo implica la producción de ávidas sondas de proteína recombinante que representan el ectodominio de los receptores unidos a la membrana. Se sabe que estos receptores conservan con frecuencia sus funciones de unión extracelular cuando sus ectodominios se expresan como una proteína soluble recombinante1. Para una proteína de interés, las proteínas recombinantes solubles se pueden producir en cualquier sistema de expresión de proteínas eucariotas adecuado en cualquier formato, siempre que se puedan oligomerizar para una mayor avidez de unión, y contiene etiquetas que se pueden utilizar en ensayos de unión basados en citometría de flujo basado en fluorescencia (por ejemplo, etiqueta FLAG, etiqueta de biotina). Los protocolos detallados para la producción de ectodominios solubles de receptores de membrana utilizando el sistema de expresión de proteínas HEK293, así como diferentes técnicas de multimerización y las construcciones de expresión de proteínas para la producción de proteínas pentaméricas y monoméricas se han descrito previamente1,,17. El protocolo aquí describirá los pasos para generar sondas fluorescentes ávidas a partir de proteínas biotiniladas monoméricas conjugandolas con estreptavidina conjugada con un fluorocromo (por ejemplo, fitoerytrina o PE), que se pueden utilizar directamente en ensayos de unión basados en células y tiene la ventaja de no requerir un anticuerpo secundario para la detección. Los protocolos generales para realizar pantallas de escala genómico ya se han descrito20,21, por lo tanto, el protocolo se centra principalmente en los detalles de la realización de pantallas de unión a proteínas recombinantes basadas en citometría de flujo utilizando el sistema de cribado knockout CRISPR/Cas9 utilizando la biblioteca Human V1 ("Yusa")18.
Protocolo
1. Producción y purificación de proteínas biotiniladas de Sus-etiquetadas
- Utilice un sistema de expresión proteica basado en células de mamíferos o insectos para producir proteínas biotiniladas recombinantes solubles (ver construcciones de plásmido en la Tabla 1).
NOTA: Kerr et al.17describe un protocolo detallado para la producción de biotina monomérica y proteínas etiquetadas con el sistema de expresión celular HEK293. Los ectodominios proteicos expresados mediante el sistema de expresión HEK293 se secretan en el medio de cultivo. - Recoger las proteínas solubles mediante la peletización de las células por centrifugación a 3.000 x g durante 20 min.
- Filtrar el sobrenadante a través de un filtro de 0,22 m y añadir las perlas de agarosa Ni2+-NTA al sobrenadante de proteína filtrada en una proporción de 1:1.000 (es decir, 50 l de 50% de purines de agarosa en 50 ml de sobrenadante). Incubar durante la noche o al menos 4-5 h a 4oC en una plataforma giratoria.
- Lave la columna de polipropileno añadiendo 5 ml de tampón de lavado de su purificación. Consulte la Tabla 2 para todas las composiciones de búfer.
- Vierta toda la mezcla sobrenadante de proteína de cordón en la columna. Las cuentas se acumularán en la base.
- Lavar las perlas 2x con 15 ml de tampón de lavado. Para evitar la dilución de proteínas, extraiga cuidadosamente el tampón de lavado residual de la columna con una jeringa de 5 ml y deseche.
- Añadir cuidadosamente 300-500 ml de tampón de su purificación directamente a las perlas e incubar durante al menos 1 h. Recoger la proteína eluida extrayendo de nuevo cuidadosamente el líquido usando una jeringa de 1 ml. Intercambie el búfer de elución al búfer deseado (por ejemplo, normalmente PBS o HBS) utilizando columnas de desaladuración. Conservar todas las proteínas a 4oC hasta su uso posterior.
2. Cuantificación y oligomerización de proteína biotinilada monomérica
NOTA: Para aumentar la avidez de unión, oligomerice las proteínas monoméricas biotiniladas en estreptavidina-PE tetramérica antes de usarlas en ensayos de unión. Lograr proporciones óptimas de conjugación de proteínas monoméricas y estreptavidina-PE tetraméricas probando una serie de dilución de monómeros biotinilados contra una concentración fija de estreptavidina y estableciendo empíricamente la dilución mínima en la que no se puede detectar un exceso de monómeros biotinilados.
- Realizar al menos ocho diluciones en serie de muestras de proteína biotinilada utilizando un tampón de dilución adecuado (YA sea PBS o HBS con albúmina sérica bovina al 1% [BSA]) en una placa de 96 pozos. Asegúrese de que el volumen final de cada dilución sea de al menos 200 l.
- Haga una placa duplicada de las muestras retirando 100 ml de cada pozo y transfiriendo a una nueva placa de 96 pozos. Incluya siempre un control. En este caso, los controles son proteínas solo etiquetas (es decir, proteínas De dominio Cd4 3+4 con etiqueta biotinilada). Esto se utilizará como sondeo de control en todos los ensayos de enlace.
- Diluir estreptavidina-PE a 0,1 g/ml en el tampón de dilución.
- En una sola de las placas, añada 100 l de la estreptavidina-PE diluida. La placa duplicada servirá como control. Añadir 100 l de tampón de dilución en la placa de control para igualar los volúmenes.
- Incubar durante 20 min a temperatura ambiente (RT). Mientras tanto, bloquee los pocillos de una placa recubierta de estreptavidina con el tampón de dilución durante 15 min.
- Transfiera el volumen total de la muestra de ambas placas a pozos individuales de las placas recubiertas de estreptavidina e incubar durante 1 h en RT.
- Lavar la placa 3x con 200 ml de tampón de lavado (es decir, PBS o HBS con 0,1% Tween-20, 2% BSA). Añadir 100 l de 2 g/ml de ratón anti-rata Cd4d3+4 IgG (OX68) e incubar durante 1 h en RT.
- Lave la placa 3x con el tampón de lavado. Añadir 100 l de un conjugado de fosfatasa alcalina antiratón a 0,2 g/ml durante 1 h en RT.
- Lavar la placa 3x con tampón de lavado y 1x en tampón de dilución.
- Preparar fosfato p-nitrofenil a 1 mg/ml en tampón de dietanolamina. Añadir 100 l en cada pocillos e incubar durante 15 min.
- Tome las lecturas de absorbancia a 405 nm. Utilice la dilución mínima en la que no hay señal en la placa como el factor de dilución adecuado para crear tetrámeros (Figura 2).
- Haga una solución de tinción de tetrórico 10x para todas las muestras y controles mediante la incubación de estreptavidina-PE de 4 g/ml y la dilución adecuada de proteínas biotiniladas durante 30 minutos en RT. Almacene las proteínas conjugadas en un tubo protegido contra la luz a 4 oC hasta su uso posterior.
3. Ensayos de unión celular basados en citometría de flujo
- Para las células adherentes, retire los medios de cultivo y lave 1x con PBS sin cationes divalentes. A continuación, agregue soluciones de desprendimiento de celdas (por ejemplo, EDTA). Deje que las células se desprendan durante 5-10 min. Toque suavemente el matraz para liberar las celdas.
NOTA: Evite el uso de productos a base de trippsina, ya que pueden cortar las proteínas de la superficie celular. - Recoger células separadas en un tubo. Para las células que crecen en suspensión (por ejemplo, células HEK293), recogen directamente las células de los matraces de cultivo en un tubo.
- Células de pellets a 200 x g durante 5 min. Retire el sobrenadante y resusppend el pellet en tampón de lavado (es decir, PBS/1% BSA).
- Cuente las células utilizando un hemocitoómetro y ajuste la concentración a 2,5 x 105-1 x 106 células/ml. Aliquot 100 l de mezcla de células preparadas en una placa de 96 pocillos en U o V. Girar la placa durante 5 min a 400 x g. Retire el sobrenadante con una pipeta multicanal.
- Añadir 100 l de sondas y controles de proteínas normalizadas con una etiqueta fluorescente altamente ávida en las placas previamente preparadas con células e incubar durante 1 h a 4oC. Después de la unión durante 1 h, gire la placa a 400 x g durante 5 min.
- Retire el sobrenadante y añada 200 l de tampón de lavado (es decir, PBS/1% BSA). Mezclar bien pipeteando hacia arriba y hacia abajo.
- Pele las células por centrifugación a 400 x g durante 5 min. Repita el paso de lavado 1x. Después de dos lavados, retire completamente el sobrenadante y resusppend el pellet celular en 100 l de PBS.
- Analizar las células por citometría de flujo. Utilice el láser amarillo-verde (es decir, 561 nm) para detectar la fluorescencia de PE.
- Primero analice las células que se han manchado con la sonda de control. Basado en la distribución de la fluorescencia de PE, dibuje una puerta para la población de unión de tal manera que no más del 1% de la célula de control caiga en esta puerta.
- Analice la muestra y determine la fracción de celdas que cae en la puerta de enlace.
NOTA: Las líneas celulares que muestran una población de unión más alta se desean para las pantallas genéticas, ya que tienen una mayor relación señal-ruido. Idealmente más del 80% de las células deben caer dentro de esta puerta.
4. Determinación de las contribuciones vinculantes de los epítopos lábiles de calor y las cadenas laterales de sulfato de heparana
NOTA: La actividad de muchas proteínas es labilisa térmica, por lo que la pérdida de actividad de unión después del tratamiento térmico es alentadora. Se aconseja determinar la contribución de los glicosaminoglicanos cargados negativamente, principalmente sulfato de heparana (HS), en la unión mediadora de las proteínas recombinantes. Esto se debe a que la unión por HS en el ensayo de unión celular descrito aquí puede ser aditivo en lugar de codependiente en otros receptores19. Esto significa que la unión observada podría ser totalmente mediada por las cadenas laterales del HS de proteoglicanos de superficie celular y no por un receptor específico. La unión al SA en la superficie celular no es necesariamente inespecífica, sino más bien una propiedad de una proteína, que es útil saber antes de realizar una pantalla genética completa.
- Preparar muestras de proteínas tratadas térmicamente para usarlas en ensayos de unión.
- Calentar la proteína monomérica normalizada pero no jugada a 80 oC durante 10 min.
- Conjugar la proteína tratada térmicamente a estreptavidina-PE asumiendo la misma relación de conjugación que su contraparte no tratada según lo determinado por ELISA (consulte la sección 2).
- Preparar muestras de proteínas bloqueadas por heparina.
- Preparar ocho diluciones 1:3 de heparina soluble en PBS con una concentración inicial de 2 mg/ml y un volumen final de 100 l.
- Incubar 100 l de sondas de unión preparadas en las diluciones de heparina durante al menos 30 min.
- Utilice proteína tratada térmicamente y los 200 ml completos de mezcla de heparina/proteína en los ensayos de unión descritos en la sección 3. Los resultados representativos se muestran en la Figura 3A,B.
5. Selección de líneas celulares que expresan establemente Cas9
NOTA: Antes de que la línea celular que une la sonda de interés se pueda utilizar en el cribado CRISPR, primero debe diseñarse para expresar la nucleasa Cas9 y un clon altamente activo seleccionado19.
- Utilice el siguiente protocolo general de producción de lentivirus para producir lentivirus utilizando la construcción lentiviral para la expresión Cas9 (consulte la Tabla 1).
- Cultivo de células HEK293-FT en medios DMEM/10% FBS a 37oC y 5% CO2. Seed HEK293-FT células 1 día antes de la transfección de modo que son -80% confluentes el día de la transfección.
NOTA: Las células HEK293FT son ligeramente adherentes; por lo tanto, cuando se utilizan para la producción de lentivirus, considere la posibilidad de encogiéndolos en frascos de cultivo recubiertos con 0,1% (p/v) gelatina para aumentar la adherencia. - Realizar transfecciones por la mañana. Añada vector de transferencia, mezcla de embalaje y reactivo de transfección a medios compatibles con la transfección preadmanido (por ejemplo, Opti-MEM). Mezclar invirtiendo el tubo 10-15x. Incubar durante 5 min en RT. Consulte la Tabla 3 para ver los volúmenes exactos.
- Agregue el reactivo de transfección como sugiere el fabricante. Mezclar por vórtices rápidos. Incubar durante 30 min en RT.
- Aspirar con mucho cuidado el medio gastado. Añada medios compatibles con la transfección a la placa.
- Agregue los complejos de reactivo/ADN de transfección por gotas en el lado de la placa y extiéndalos lentamente a través de la placa girando muy suavemente.
- Incubar a 37oC durante 3-5 h y sustituir el medio por el medio D10. Incubar durante la noche.
- Al día siguiente por la mañana, sustituya el medio por un medio fresco D10. Incubar durante la noche.
- Al día siguiente, tarde en la tarde, recoger el sobrenadante viral. Filtrar con un filtro de 0,45 m con baja unión a proteínas. Opcionalmente, añadir el medio D10 fresco, incubar durante la noche y recordar el sobrenadante al día siguiente.
- Los sobrenadantes de virus son estables a 4 oC durante unos días. Almacenar a -80 oC para almacenamiento a largo plazo.
NOTA: Para generar una preparación lentiviral altamente concentrada, que podría ser deseable para la transducción de células difíciles de transducir, los sobrenadantes también se pueden concentrar por centrifugación a 6.000 x g durante la noche a 4 oC. Marque el pellet viral translúcido con una pluma resistente al etanol y deseche el sobrenadante. Resuspender el pellet en 1/100 del volumen original para un aumento de 100x en la concentración.
- Cultivo de células HEK293-FT en medios DMEM/10% FBS a 37oC y 5% CO2. Seed HEK293-FT células 1 día antes de la transfección de modo que son -80% confluentes el día de la transfección.
- Transducir las células con lentivirus.
- Placa 1 x 106 células por pozo en una placa de 6 pozos con 3 ml de medios de cultivo apropiados. Algunas células son más fácilmente transducidas que otras. Para células fáciles de transducir (por ejemplo, células HEK), agregue directamente el lentivirus a las células. Para las células difíciles de transducir, podría ser necesario seguir un protocolo de espinculación como se describe a continuación.
- Alícuota 2 ml de 2-5 x 106 células/ml en un tubo cónico de 15 ml.
- Añadir lentivirus junto con bromuro de hexadimetrina de 8 g/ml e incubar a RT durante 30 min.
- Centrífuga durante 100 min a 800 x g a 32oC. Luego resuspendió las células en el mismo medio y agregue la suspensión celular en matraces de cultivo apropiados con medios apropiados.
- Permitir transducciones durante al menos 24 h. Después retire el medio que contiene el virus y agregue un medio fresco.
- Después de otras 24 h, cambie los medios a uno que se complemente con los antibióticos adecuados. La construcción Cas9 contiene un casete de resistencia a blasticidina para su selección.
NOTA: La cantidad de blasticidina debe optimizarse para cada línea celular realizando una curva de muerte de respuesta a la dosis. Una concentración de blasticidina entre 2,5-50 g/ml debe matar la mayoría de las líneas celulares no transducidas dentro de los 10 días posteriores a la transducción.
- Placa 1 x 106 células por pozo en una placa de 6 pozos con 3 ml de medios de cultivo apropiados. Algunas células son más fácilmente transducidas que otras. Para células fáciles de transducir (por ejemplo, células HEK), agregue directamente el lentivirus a las células. Para las células difíciles de transducir, podría ser necesario seguir un protocolo de espinculación como se describe a continuación.
- Realizar la selección hasta que se maten todas las células de la placa de control (es decir, las células no traducidas que han sido tratadas con la misma concentración de antibióticos de selección).
6. Selección de clones de alta actividad Cas9
NOTA: Polyclonal Cas9 se puede utilizar para realizar con éxito pantallas genéticas; sin embargo, la selección de un clon con alta actividad Cas9 mejora los resultados de cribado18.
- Utilice la dilución limitante o las células individuales resistentes a blasticidinas de una sola célula en pozos de tres placas de 96 pozos que contienen medios de cultivo complementados con blasticidina. Los clones comenzarán a emerger entre 2-4 semanas. Seleccione 10-20 clones y expándalos en 6 placas de pozo.
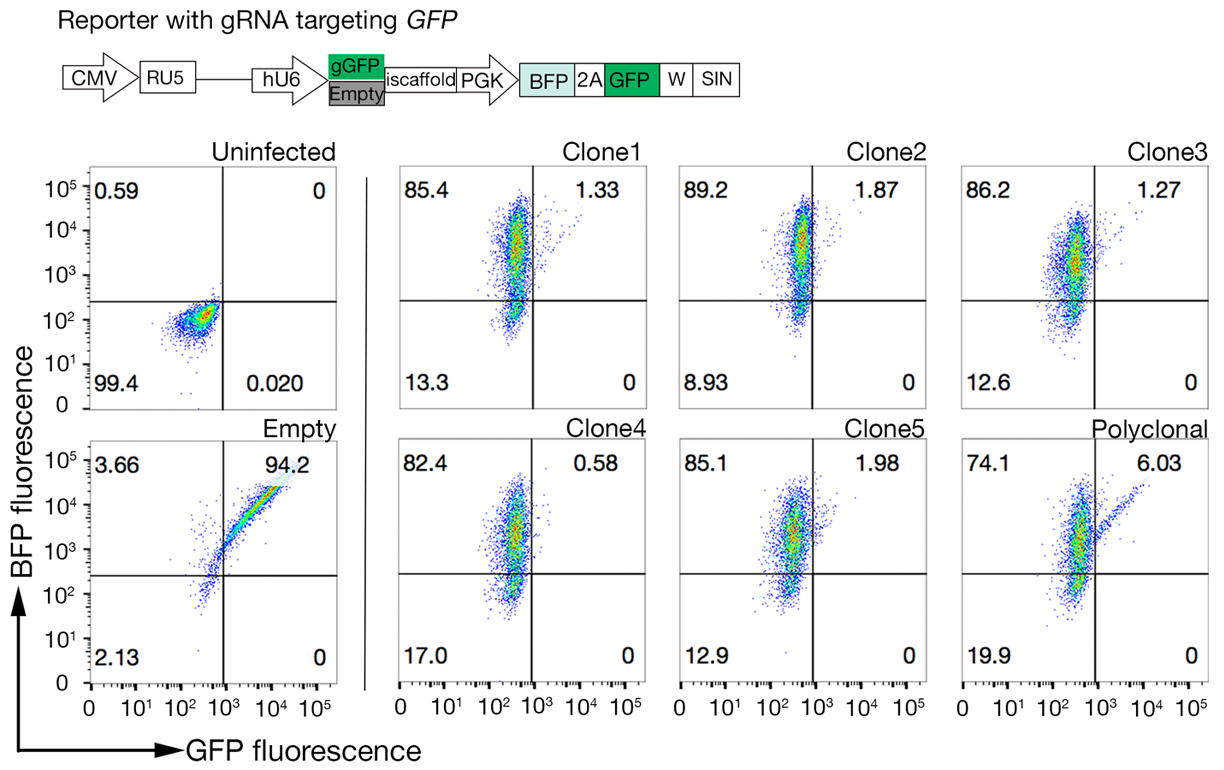
- Ensayo de los clones para la actividad cas9 utilizando el sistema de evaluación rápida GFP-BFP (proteína fluorescente verde-proteína azul fluorescente), que utiliza un sistema de eliminación de génicas exógenas en el que las células se trans inducen ya sea con una construcción que expresa la GFP con un GRNA orientado a la GFP o un GRNA vacío como control18.
- Ordene plásmidos de reportero: Plásmido GFP-BFP, Plásmido Control-BFP (Tabla 1).
- Produzca lentivirus tanto para el plásmido GFP-BFP como para el plásmido Control-BFP utilizando el protocolo de producción de lentivirus descrito en la sección 5.1.
- Transducir cada clon de línea celular que expresa Cas9 con el lentivirus que codifica el sistema GFP-BFP y El Control-BFP por separado. Siga el protocolo de la sección 5.2.
- Después de 3 días de transducción, examine la fluorescencia GFP-BFP de cada clon utilizando citometría de flujo. Utilice láser de 488 nm y láser de 405 nm para detectar GFP y BFP respectivamente.
- Custete la actividad Cas9 en cada clon examinando la relación de BFP solo con las celdas positivas dobles GFP-BFP. Idealmente, las células Cas9 de alta actividad deberían tener >95% de eficiencia de eliminatorias de GFP(Figura 4).
7. Generación de la biblioteca de selección de knockout CRISPR-Cas9 en todo el genoma
- Para el cribado en todo el genoma utilizando la biblioteca Human V118, pida la biblioteca de todo el genoma (consulte la Tabla 1)y prepare la biblioteca de plásmidos a partir de la puñalada bacteriana utilizando el protocolo proporcionado en "Protocolos para la replicación de bibliotecas" en el manual del fabricante.
- Utilice la preparación del plásmido de la biblioteca de todo el genoma para producir una biblioteca lentiviral que codifica los gRNAs para la interrupción específica de los genes humanos utilizando el protocolo de producción de lentivirus descrito en la sección 5.1.
NOTA: Una buena práctica es producir un solo lote de preparación lentiviral que esté optimizado para la transducción para mejorar la consistencia experimental. - Utilice el protocolo de transducción de la sección 5.2 para realizar transducciones de prueba a pequeña escala para determinar la cantidad necesaria de virus para cada línea celular para lograr una transducción del 30%. Utilice la citometría de flujo para evaluar la fluorescencia de BFP como un proxy para la eficiencia de la transducción.
- Para transducir las células HEK293, simplemente agregue la preparación lentiviral predeterminada a 30-50 x 106 células cultivadas en medios de crecimiento normal durante 4 h. A continuación, retire los medios con lentivirus y sustitúyalo por medios de crecimiento frescos.
- Para otras líneas celulares, utilice el protocolo de espinculación en la sección 5.2.1 pero a una escala mayor, de tal manera que se transduzca un total de 30-50 x 106 celdas. Para ello, alícuota 2 mL de 5 x 106 células/ml en un tubo cónico de 15 ml y proceda como se indica.
- Para las líneas celulares adherentes, seleccione las células transducidas agregando puromicina 24 h después de la transducción.
NOTA: Optimice las concentraciones de puromicina realizando una curva de eliminación de respuesta a la dosis. Normalmente, las concentraciones entre 1-10 g/ml deben matar las células no traducidas en un plazo de 3-5 días. Evitar el uso de concentraciones más altas de puromicina porque esto puede aumentar las posibilidades de seleccionar las células que han sido transducidas por más de ARN guía único (sgRNA). - Para las células de suspensión, cosecha las células transducidas (es decir, BFP positivas) 3 días después de la transición utilizando un clasificador de células y genera bibliotecas que contienen al menos 10 x 106 celdas. Una vez seleccionadas con BFP, haz crecer las células en medios complementados con la cantidad adecuada de puromicina.
NOTA: Evite las selecciones solo con puromicina para las líneas celulares de suspensión, ya que es difícil eliminar las células muertas y los desechos de los cultivos celulares de suspensión que pueden interferir con la clasificación celular. - Biblioteca mutante de la cultura durante 9-16 días después de latransducción con paso regular cada 2-3 días.
8. Examen genético para la unión de superficies celulares
- Pele la biblioteca de celdas mutantes a 200 x g durante 5 minutos y resuspendir las células en PBS.
- Divida las células en dos tubos cónicos de 15 ml con al menos 50 x 106 células en cada tubo.
- Gire un tubo cónico a 200 x g durante 5 min, retire el sobrenadante y congele el pellet celular a -20 oC. Esta es la población de control y se procesará más adelante.
- Resuspender el pellet en el otro tubo en 10 ml de PBS/1% BSA. Reservar 100 l de células como control negativo en una placa de 96 pozos.
- Agregue la proteína recombinante preconjugada apropiada a la suspensión celular en el tubo cónico y las proteínas de control negativo a la placa de 96 pozos.
- Realizar la tinción celular durante al menos 1 h a 4 oC en un rotor de sobremesa con rotación suave (6 rpm).
- Pele el de las células a 200 x g durante 5 min, retire el sobrenadante. Realice dos pasos de lavado, luego resuspender las células en 5 ml de PBS.
- Tensar las células a través de un colador de células de 30 m para eliminar los racimos de células. Analizar utilizando un clasificador de flujo.
- Utilice la muestra de control negativo para gate para celdas BFP+/PE-.
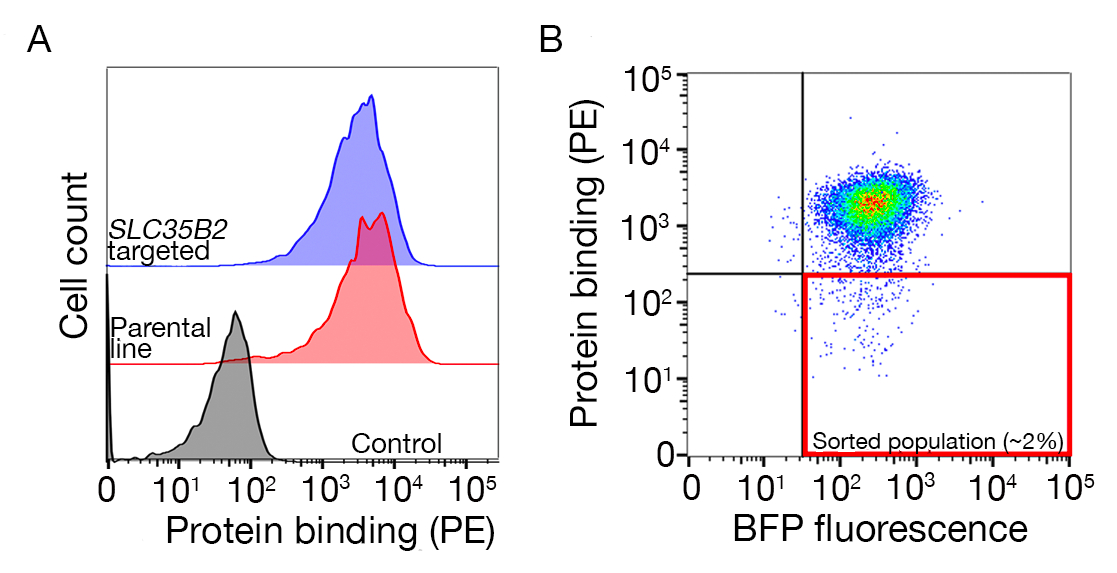
- Ordene la muestra y recoja las celdas BFP+/PE-. Las puertas de clasificación dependerán de la unión de las células a la proteína, pero normalmente se recogen el 1-5% de las muestras negativas de PE. Un ejemplo de una puerta de clasificación se proporciona en la figura suplementaria 1.
- Recoja 500,000-1,000,000 celdas de la puerta seleccionada. Dado el bajo número de células, considere la posibilidad de recoger las muestras en un tubo de centrifugación de 1,5 ml para minimizar las pérdidas.
- Pele el pelado de las células ordenadas centrifugando a 500 x g durante 5 min. Retire cuidadosamente el sobrenadante y deseche. Es posible almacenar el pellet a -20 oC durante un máximo de 6 meses.
9. Extracción de ADN genómico y primera PCR para el enriquecimiento del ARNN
- Extraiga ADN genómico de la población de control de alta complejidad.
- Si la población de control se congeló a -20 oC, saque el tubo cónico y agregue PBS. Mantener sobre hielo para descongelar el pellet.
- Utilice un kit comercial (ver Tabla de Materiales)utilizando las recomendaciones del fabricante para extraer ADN genómico de 50 x 106 células. Ajuste la concentración de ADN a 1 mg/ml.
- Para cada muestra, configure una mezcla maestra para PCR correspondiente a 72 g de ADN. Alícuota 50 l por pozo en 36 pocillos de una placa PCR de 96 pozos. Las secuencias de imprimación necesarias se enumeran en la Tabla 4. Utilice la guía de las Tablas 5 y 6.
- Resuelva 5 l de la PCR a partir de 6-12 muestras representativas en un gel de agarosa del 2% (p/v). Se debe observar una sola banda transparente a 250 bp. Si las bandas son débiles, repita el PCR durante 2-3 ciclos adicionales.
- Utilice una pipeta multicanal para recoger 5 l de productos de PCR de cada pocero (180 ml en total) y agruparlos en un depósito con 900 ml de tampón de unión de un kit comercial (ver Tabla de materiales).
- Purifique los productos PCR utilizando un kit comercial de purificación de PCR. Elte el ADN en 50 ml de tampón de elución de un kit comercial (ver Tabla de Materiales) y mida la concentración de ADN.
- Es poco probable que las muestras que se han clasificado para la pérdida de fenotipo de unión se componen de un gran número de clones independientes. Por lo tanto, no es necesario realizar la PCR con 72 g de ADN. Aísle el ADN utilizando un kit comercial adecuado (ver Tabla de materiales). Configure 3-4 reacciones de PCR utilizando el protocolo descrito anteriormente (sección 9.1.3) con ADN de 100 ng/L. Si el número de celda ordenado es inferior a 100.000, utilice lysates celulares en lugar de preparaciones de ADN genómico.
- Aliquot aproximadamente 10.000 células/pozo en una placa PCR de 96 pozos.
- Pele el pelado de las células en la placa y retire cuidadosamente la mayor parte del sobrenadante. El pellet no será visible.
- Añadir 25 l de agua en cada pocótela y calentar las muestras a 95oC durante 10 min.
- Añadir 5 l de 2 mg/ml de proteínaasa recién diluida K a cada pocóter durante 1 h e incubar a 56oC. A continuación, calentar la muestra durante 10 min a 95 oC para inactivar la proteinasa K.
- Utilice 10 l de mezcla de izado celular por reacción PCR. Los litos deben utilizarse en un plazo de 24 h.
10. Segunda ronda de PCR para la codificación de barras de índice y la secuenciación
- Diluir los productos desde la PCR de primera ronda hasta 40 pg/L.
- Configure un PCR por muestra (utilice la guía proporcionada en los cuadros 7 y 8). El uso de una polimerasa de alta fidelidad es importante para minimizar los errores introducidos por la polimerasa durante la amplificación de sgRNA.
- Resuelva 5 l de productos de PCR en un gel de agarosa del 2% (p/v). Se debe observar una sola banda transparente a 330 bps.
- Purifique los productos de PCR utilizando perlas paramagnéticas añadiendo 31,5 l de (0,7 veces el volumen total) de perlas resuspendidas a los productos de PCR, mezclando bien e incubando durante 5 minutos en RT.
- Coloque el tubo en un bastidor magnético durante 3 minutos. Las perlas deben ser capturadas en el lado de la placa y la solución debe ser clara. Retire y deseche cuidadosamente el sobrenadante.
- Añadir 150 l de etanol 80% recién preparado al tubo. Incubar durante 30 s, y luego retirar y desechar cuidadosamente el sobrenadante.
- Repita el paso 13.6, esta vez con 180 l. A continuación, seque al aire las cuentas durante 5 minutos.
- Retire el tubo del imán. Elute objetivo de ADN de cuentas a 35 ml de tampón de EB estéril. Incubar durante 3 min, luego volver a colocar el tubo en el bastidor magnético durante 3 min.
- Transfiera aproximadamente 30 l del sobrenadante que contiene los productos de PCR eluidos a un tubo limpio.
- Secuenciar los ejemplos en una plataforma de secuenciación de próxima generación. Para la biblioteca de GRNA HumanV1, utilice la imprimación personalizada que se muestra en la Tabla 4 para secuenciar 19 bp.
11. Análisis bioinformático para identificar el receptor y las vías conexas
- Asigne secuencias de población ordenada y no ordenada a la biblioteca de referencia mediante la función de recuento de MAGeCK. La función producirá un archivo de recuento sin procesar(Tabla complementaria 1).
NOTA: Las instrucciones detalladas sobre la instalación de MAGeCK y el uso de diferentes funciones dentro de MAGeCK se describen en un protocolo publicado previamente por Wang et al.20. - Compruebe el estándar técnico de la biblioteca de control utilizada en la pantalla.
- Mediana normalizar los recuentos sin procesar y utilizar el paquete ggplot2 en R21 o software equivalente para trazar una gráfica de función de densidad acumulativa empírica de los recuentos en plásmido y controlar muestras no ordenadas (Figura 5A).
- Ejecute la función -test de MAGeCK utilizando recuentos de población de plásmidos como "control" y los recuentos de muestras de control no ordenadas como la muestra de "prueba". La función es producirá un archivo de resumen genético(Tabla suplementaria 1).
- Abra el archivo de resumen del gen y dibuje la distribución de log-fold-changes (columnaneg-lfc) para los genes esenciales y no esenciales previamente categorizados22 (Figura 5B).
- Seleccione genes significativamente agotados(neg-fdr < 0.05) y realice análisis de enriquecimiento de vías utilizando el paquete de enriquecimiento23 o cualquier paquete de enriquecimiento de vía equivalente en R (Figura 5C).
- Ejecute la función -test de MAGeCK con la configuración predeterminada. Utilice recuentos sin procesar de la muestra de control no ordenada como "control" y cuenta desde la muestra ordenada como "tratamiento" al realizar el análisis.
- Abra el archivo de resumen de genes generado por MAGeCK y clasifique la columna pos-rank en orden ascendente. Utilice FDR ( columnapos-fdr) < 0.05 como un límite para la identificación de los hits. El receptor se clasifica generalmente altamente, a menudo en la primera posición.
- Trazar las puntuaciones de Robust-Ranking-Algorithm (RRA) para la selección positiva (pos-score) en R o en un software equivalente (Figura 5D).
- Seleccione los golpes genéticos y realice análisis de enriquecimiento de vías utilizando el paquete de enriquecimiento o cualquier paquete de enriquecimiento de vías equivalentes en R para identificar las vías enriquecidas.
Resultados
Se proporcionan datos de secuenciación de dos pantallas nocaut a escala del genoma representativas para la identificación del socio vinculante de las células humanas TNFSF9 y P. falciparum RH5 realizadas en células NCI-SNU-1 y HEK293 respectivamente (Tabla complementaria 1). El comportamiento de unión de RH5 se vio afectado tanto por el sulfato de heparana como por su receptor conocido BSG24 (Figura 3C), mientras que TNFRSF9 se une específicamente a su receptor conocido TNFSF9 y no perdió la unión al preincubación con heparina soluble. La proteína 3 de la Figura 3B representa TNFRSF9.
Para ambas líneas celulares, también se proporciona la distribución de gRNAs en la biblioteca mutante de control después de 3 días (9, 14 y 16 días después de la posteriortransducción)(Tabla complementaria 1). La distribución del ARNN reveló que la complejidad de la biblioteca se mantuvo durante el transcurso del experimento(Figura 5A). La prueba genética para la identificación del ligando para TNFSF9 se realizó el día 14 después de la posttransducción, mientras que para RH5 se realizó el día 9 después de la trasducción. La calidad técnica de las pantallas se evaluó examinando la distribución de los cambios de pliegue observados de los gRNAs dirigidos a un conjunto de referencia de genes no esenciales en comparación con la distribución del conjunto de referencia de genes esenciales22 (Figura 5B). Además, el enriquecimiento a nivel de vía también reveló que las vías esenciales esperadas fueron identificadas y enriquecidas significativamente en la población "deserción" al comparar la muestra de control con la biblioteca original de plásmidos. Un ejemplo con el día 14 NCI-SNU-1 muestra se representa en el cuadro 5C.
La distribución de los gRNAs en el control frente a la población ordenada utilizando la función -test de MAGeCK (ver Tabla Suplementaria 1 para la salida de resumen genético de MAGeCK) se utilizó para identificar el receptor de las pantallas fenotípicas. La puntuación RRA modificada notificada por MAGeCK en el análisis a nivel genético se traza contra los genes clasificados por valores p. La puntuación RRA en MAGeCK proporciona una medida en la que los gRNAs se clasifican consistentemente más alto de lo esperado. En la pantalla de TNFRSF9, el mayor éxito fue TNFSF9, que es un socio de enlace conocido de TNFRSF9 (Figura 5D). Además, también se identificaron varios genes relacionados con la vía TP53. En el caso de RH5, además del receptor conocido (BSG) y el gen necesario para la producción de los GAG sulfatados (SLC35B2), también se identificó un gen adicional (SLC16A1) (Figura 5E). SLC16A1 es un acompañante necesario para el tráfico de BSG a la superficie de las células25. Juntos, estos resultados demuestran la capacidad de la pantalla para identificar los receptores que interactúan directamente y los componentes celulares necesarios para que ese receptor se exprese en la superficie de las células en una forma funcional.

Figura 1: Visión general del enfoque de cribado genético para identificar los receptores de la superficie celular. Este ensayo consta de tres pasos principales: En primer lugar, las proteínas recombinantes que representan el ectodominio de los receptores de superficie celular se expresan en una línea celular que puede agregar modificaciones posttranslacionales estructuralmente críticas como las células HEK293. Los ectodominios de proteína monomérica se oligomerizan conjugando con streptavidin-PE para aumentar su avidez de unión. En segundo lugar, estas ávidas sondas se utilizan en ensayos de unión celular donde la tinción brillante en las líneas celulares indicada por un cambio prominente en la fluorescencia de PE (en verde) en comparación con una proteína de control negativo (en negro) demuestra la presencia de un socio de unión a la superficie celular. En tercer lugar, se seleccionan las líneas celulares que expresan Cas9 positivas como receptores y se realiza un cribado a escala del genoma utilizando gRNAs dirigido a la gran mayoría de los genes codificantes de proteínas. Al generar bibliotecas mutantes, es común utilizar una eficiencia de transducción del 30%, que se basa en la probabilidad de distribución de Poisson que garantiza que cada célula reciba un solo ARN de tal manera que el fenotipo resultante se atribuya a un nocaut específico. El marcador BFP expresado por las celdas transducidas se utiliza para seleccionar celdas que contienen gRNAs mediante FACS. Las pantallas fenotípicas se realizan entre 9-16 días después de latransducción. El día de la pantalla, la población total de células mutantes se divide en dos. La mitad se mantiene como la población de control y la otra mitad se selecciona para la unión a proteínas recombinantes. Las células de la biblioteca mutante que ya no son capaces de unir la proteína recombinante se ordenan mediante FACS y el enriquecimiento de los gRNAs en la población ordenada frente a la de control se utiliza para identificar los genes necesarios para la unión a la superficie celular de la sonda ávida etiquetada. Se indican los pasos en el protocolo que requieren un tiempo considerable. Esta cifra ha sido modificada de Sharma et al.19. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Establecimiento de las proporciones de proteína biotinilada a streptavidin-PE utilizando un método basado en ELISA. Un ejemplo de estrategia de conjugación estreptavidina-PE utilizada para generar una sonda ávida a partir de una proteína monomérica biotinilada. Se incuba una serie de dilución de monómeros biotinilados contra una concentración fija de estreptavidina. ELISA determinó la dilución mínima en la que no se pueden detectar monómeros biotinilados en exceso. ELISA se realizó con o sin preincuentar una gama de diluciones proteicas con 10 ng de estreptavidina-PE. En presencia de estreptavidina-PE, se calificó la dilución mínima en la que no se identificó ninguna señal (negro en círculo) y la cantidad de proteína necesaria para la saturación para generar una solución de 10x en stock con 4 g/ml de streptavidin-PE. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Unión representativa de proteínas a líneas celulares. (A) La unión de proteínas a las líneas celulares tuvo un claro aumento de la fluorescencia asociada a la célula en comparación con la muestra de control. El tratamiento térmico (80 oC durante 10 min) de proteína recombinante abroinó toda la unión a un control negativo, demostrando que el comportamiento de unión dependía de la proteína correctamente doblada. (B) Diferentes clases de comportamiento de unión de proteínas a superficies celulares; dependencia de los GAG. De izquierda a derecha, las proteínas se pueden clasificar en tres tipos: La proteína tipo 1 sólo se adsorbe al HS. Estas proteínas pierden su unión después de la preincubación con concentraciones de heparina superiores a 0,2 mg/ml. La proteína tipo 2 se une al HS además de un receptor específico. Estas proteínas pierden la unión parcial en los experimentos de prebloqueo. La proteína tipo 3 no une el HS. Estas proteínas no pierden la unión en comparación con las líneas parentales. (C) Un ejemplo de una proteína (es decir, RH5) que se une al SE Y y a un receptor específico de forma aditiva. Dirigirse al receptor (es decir, BSG) o a las enzimas necesarias para la síntesis del HS (por ejemplo, SLC35B2, EXTL3) sólo reduce parcialmente la unión de RH5 a las células en relación con los controles. Las líneas policonales transducidas se pueden utilizar en tales experimentos para establecer el comportamiento de unión. Esta cifra ha sido modificada de Sharma et al.19. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Selección de líneas celulares clonales con alta actividad Cas9. La eficiencia de edición del genoma de las líneas policlonales y clonadas de las líneas celulares NCI-SNU-1 se evaluó utilizando el sistema de reportero GFP-BFP, en el que las líneas celulares se transducieron con virus con un plásmido de GFP codificado con ARNN o sin (es decir, "vacío"). Se representa un esquema. La citometría de flujo se utilizó para probar la expresión de BFP y GFP después de la transducción y en comparación con el control no infectado. La expresión GFP se utilizó como proxy para la actividad Cas9, mientras que la expresión BFP marcó celdas transducidas. El perfil de las células infectadas no infectadas y vacías era similar para todos los clones. Los perfiles representativos se representan en el panel izquierdo. Los cinco clones de la línea celular NCI-SNU-1 mostraron una mayor pérdida de GFP en comparación con la línea policlonal (panel derecho), con el clon 4 mostrando la mayor eficiencia con la población refractaria más baja. Esta cifra ha sido modificada de Sharma et al.19. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Resultados representativos de las pruebas genéticas para la identificación de los socios de unión a la superficie celular. (A) Gráficas de función de distribución acumulativa que comparan la abundancia de ARNG en la biblioteca de plásmidos con las bibliotecas mutantes de células HEK-293-E y NCI-SNU-1 los días 9, 14 y 16 después de la posteriortransducción. Para cualquier número dado, la función de densidad acumulativa notifica el porcentaje de puntos de datos que estaban por debajo de ese umbral. El pequeño desplazamiento de la población de células mutantes en comparación con la población original de plásmidos representa el agotamiento en un subconjunto de gRNAs en comparación con la biblioteca de plásmidos. (B) Distribución de los cambios de log-fold en genes que han sido previamente clasificados como esenciales (rojo) o no esenciales (negros) en las líneas celulares HEK293 y NCI-SNU-1. La distribución de los cambios plegables para los genes no esenciales se centró en el número 0, mientras que para los genes esenciales se desplazó hacia la izquierda hacia cambios negativos de pliegue. (C) Vías significativamente enriquecidas en genes agotados en la población de control mutante NCI-SNU-1 14 días después de latransducción. Se identificaron las vías conocidas conocidas de las células esenciales. (D) Algoritmo de rango robusto (RRA)-puntuación para genes que se enriquecieron en las células ordenadas que habían perdido la capacidad de unir la sonda TNFRSF9. Los genes fueron clasificados de acuerdo con la puntuación RRA. En la pantalla se identificaron el socio de interacción conocido TNFSF9 y los genes relacionados con la vía TP53 (etiquetados en rojo). (E) Puntuaciones RRA ordenadas por rango para genes identificados a partir del análisis de enriquecimiento de ARNN requerido para la unión RH5 a células HEK293 (panel izquierdo). SLC35B2 y SLC16A1 se identificaron dentro de un umbral de tasa de descubrimiento falso (FDR) del 5%. Se identificaron dos genes adicionales en la vía de biosíntesis del SST (es decir, EXTL3 y NDST1)dentro de FDR del 25%. Esquema que representa la vía general de biosíntesis GAG con los genes relevantes asignados a los pasos correspondientes (panel 2). Los genes necesarios para el compromiso con la biogénesis de sulfato de condroitina (es decir, CSGALNACT1/2)no se identificaron en la pantalla. Esta cifra ha sido modificada de Sharma et al.19. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Nombre de Plásmido | Plásmido # | Uso |
| Construcción de la expresión proteica: CD200RCD4d3+4-bio-linker-his | Addgene: 36153 | Producción de proteína recombinante con CD4d3+4, biotina y 6 etiquetas. |
| pMD2.G | Addgene: 12259 | Sobre VSV-G que expresa plásmido; producción de lentivirus |
| psPAX2 | Addgene: 12260 | Plásmido de envasado Lentiviral, producción de lentivirus |
| Cas9-construcción: pKLV2-EF1a-Cas9Bsd-W | Addgene: 68343 | Producción de la línea Cas9 que expresa constitutivamente |
| Construcción de expresión de gRNA: pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W | Addgene: 67974 | Vector de expresión de ARNN CRISPR con un andamio mejorado y marcadores puros/BFP |
| Biblioteca CRUJIENTE de Knockout en todo el genoma mejorada del ser humano | Addgene: 67989 | Una biblioteca de ARNN contra 18.010 genes humanos, diseñada para su uso en lentivirus. |
| Construcción GFP-BFP: pKLV2-U6gRNA5(gGFP)-PGKBFP2AGFP-W | Addgene: 67980 | Reportero de actividad Cas9 con BFP y GFP. |
| Construcción vacía: pKLV2-U6gRNA5(vacío)-PGKBFP2AGFP-W | Addgene: 67979 | Reportero de actividad Cas9 (control) con BFP y GFP. |
Tabla 1: Plásmidos utilizados en este enfoque.
| Nombre del búfer | Componentes |
| HBS (10X) | 1,5 M NaCl y 200 mM HEPES en agua MiliQ, ajuste a pH 7.4 |
| PBS (10X) | 80 g NaCl, 2 g KCl, 14,4 g Na2HPO4 y 2,4 g KH2PO4 en agua MiliQ, ajustar a pH 7.4 |
| Tampón de fosfato sódico (80mM stock) | 7.1 g Na2HPO4.2H2O, 5.55 g NaH2PO4, ajustar a pH 7.4 |
| Búfer de unión a su purificación | 20 mM Tampón de fosfato sódico, 0,5 M NaCl y 20 mM de imidazol, ajustar a pH 7,4 |
| Búfer de elución de purificación | 20 mM Tampón de fosfato sódico, 0,5M NaCl y 400 mM de imidazol, ajustar a pH 7,4 |
| Tampón de dietanolamina | 10% dietanolamina y 0,5 mM MgCl2 en agua MiliQ, ajuste a pH 9.2: |
| D10 | DMEM, 1% penicilina-estreptomicina (100 unidades/ml) y 10% inactivado por calor FBS |
Tabla 2: Zonas de influencia necesarias para este estudio.
| Componentes | Plato de 10 cm | Placa de 6 pozos |
| 293 célulasFT | 70–80% confluente | 70–80% confluente |
| Medios compatibles con la transfección (Opti-MEM) (paso 5.1.2) | 3 mL | 500 l |
| Medios compatibles con la transfección (Opti-MEM) (paso 5.1.4) | 5 mL | 2 mL |
| Vector de transferencia Lentiviral | 3 g | 0,5 g |
| psPax2 (ver tabla 1) | 7.4 g | 1.2 g |
| pMD2.G (véase la tabla 1) | 1.6 g | 0.25 g |
| Reactivo PLUS | 12 l | 2 l |
| Lipofectamine LTX | 36 l | 6 L |
| D10 (Paso 7.1.7) | 5 mL | 1,5 mL |
| D10 (Pasos 7.1.8 y 7.1.10) | 8 mL | 2 mL |
Tabla 3: Cantidades y volúmenes de reactivos para la mezcla de envases de lentivirus.
Tabla 4: Secuencias de primer plano para amplificar el ARNn. Haga clic aquí para ver este archivo (Haga clic con el botón derecho para descargar).
| Reactivo | Volumen por reacción | Mezcla maestra (x38) |
| Q5 Hot Start High-Fidelity 2x | 25 l | 950 l |
| Mezcla de imprimación (L1/U1) (10oM cada una) | 1 l | 38 l |
| ADN genómico (1 mg/ml) | 2 l | 72 l |
| H2O | 22 l | 1100 l |
| Total | 50 l | 1900 l |
Tabla 5: PCR para la amplificación de gRNAs a partir de muestras de alta complejidad.
| Número de ciclo | Desnaturalizar | Recocido | Extensión |
| 1 | 98 oC, 30s | ||
| 2-24 | 98 oC, 10s | 61 oC, 15s | 72 oC, 20s |
| 25 | 72 oC, 2 min |
Tabla 6: Condiciones de PCR para el primer PCR.
| Reactivo | Volumen por reacción |
| KAPA HiFi HotStart ReadyMix | 25 l |
| Mezcla de imprimación (PE1.0/imprimación de índice) (5 m cada una) | 2 litros |
| Primer producto pcR (40 pg/l) | 5 l |
| H2O | 18 l |
| Total | 50 l |
Tabla 7: PCR para el etiquetado de índices de sgNNAs a partir de pantallas genéticas.
| Número de ciclo | Desnaturalizar | Recocido | Extensión |
| 1 | 98 oC, 30s | ||
| 2-15 | 98 oC, 10s | 66 oC, 15s | 72 oC, 20s |
| 16 | 72 oC, 5 min |
Tabla 8: Condiciones de PCR para el segundo PCR.

Figura suplementaria S1: Una guía para dibujar puertas para ordenar la población no vinculada. (A) Un candidato ideal de proteínas para el cribado debe tener un claro desplazamiento de la población de unión en comparación con la población de control y la unión debe conservarse en las células que carecen de maquinaria para la biosíntesis del SS. Un experimento de bloqueo de heparina se puede utilizar en lugar de pruebas en líneas celulares dirigidas SLC35B2. (B) Se recogieron las células que carecían de la tinción superficial del ectodominio proteico pero que expresaban fluorescencia de BFP a partir de la transducción lentiviral. Las células mostradas son de una pantalla para la identificación del receptor para GABBR222. Esta cifra ha sido modificada de Sharma et al.19. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

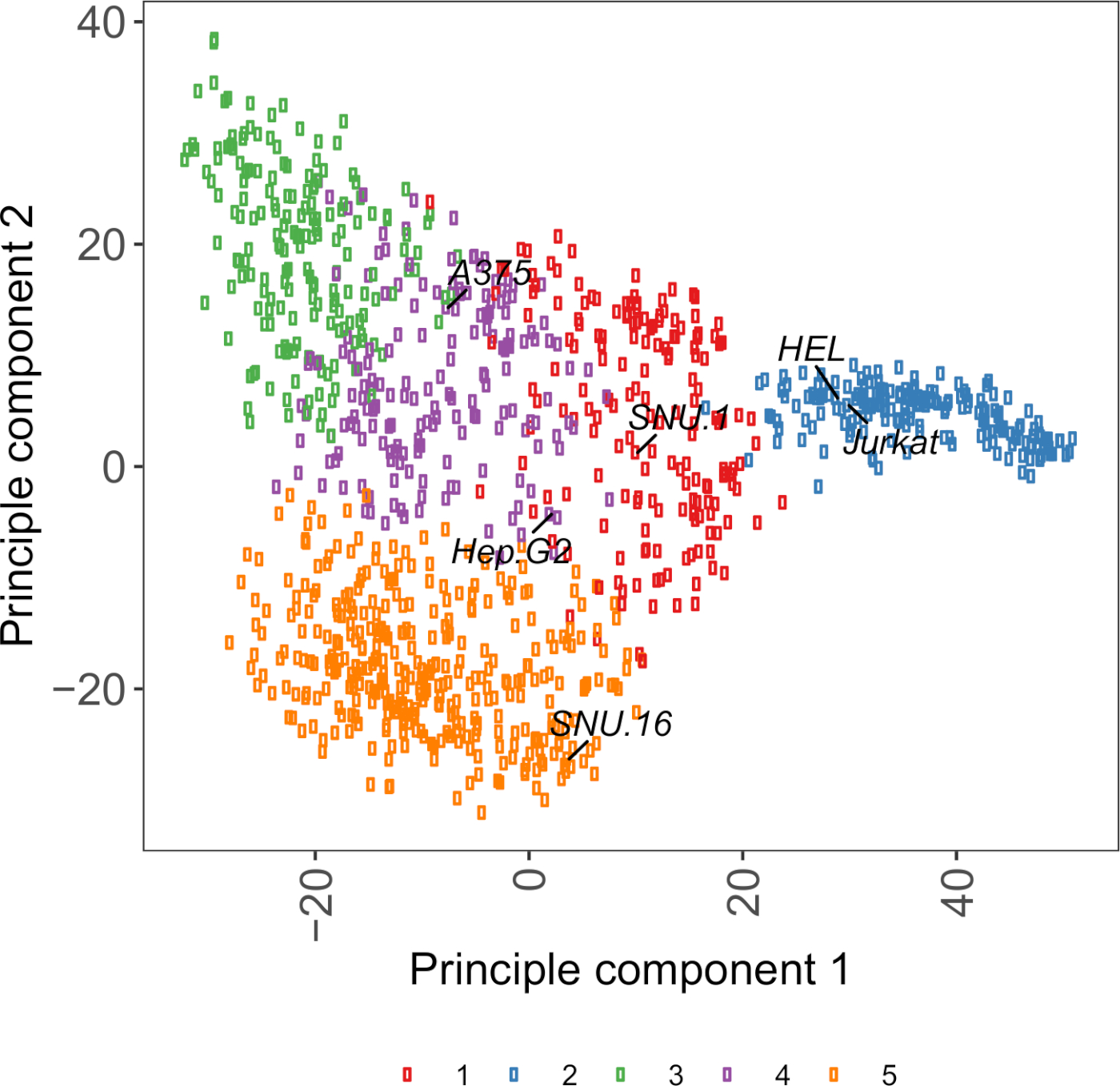
Figura suplementaria S2: Gráfica pcA basada en la transcriptocina de glicoproteína de superficie celular utilizando datos de ARN-seq de más de 1.000 líneas celulares de cáncer. Las líneas celulares de Cell Model Passport27 se agruparon utilizando clustering de medios K de acuerdo con los valores de FPKM de glicoproteínas de superficie de celda de 1.500 celdas. Las líneas de celda representativas de cada clúster están etiquetadas. El grupo 5 estaba compuesto enteramente por líneas celulares de origen hematopoyético (véase también la Tabla Suplementaria 2). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

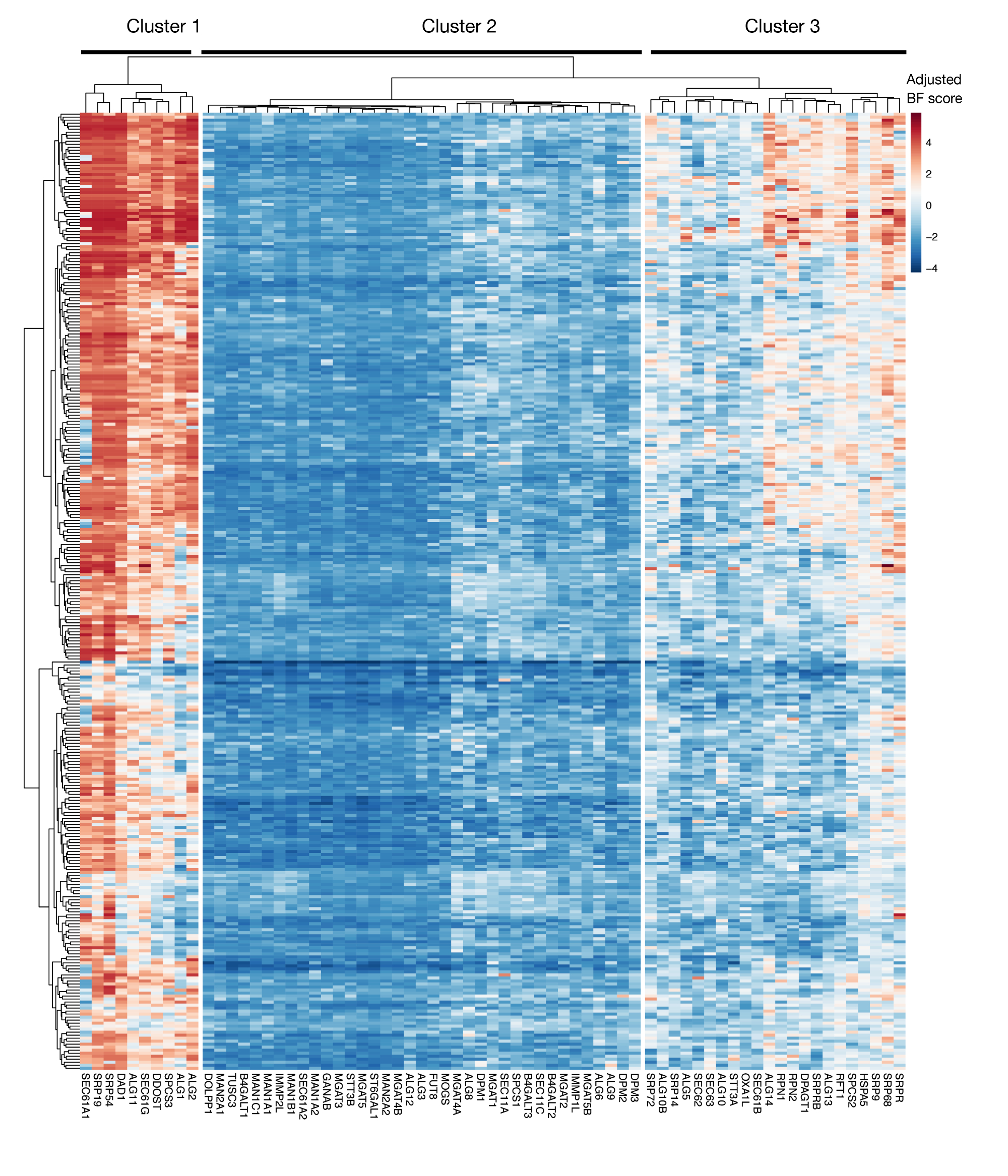
Figura suplementaria S3: Puntuaciones de esencialidad para la exportación de proteínas de anotación KEGG y genes de glicosilación ligadas a N a partir de puntuaciones de proyectos. Las puntuaciones de esencialidad de Bayes ajustadas para 330 líneas celulares (columnas, no etiquetadas) se trazan para genes de exportación de proteínas y vía de glicosilación ligada A N (eje X). Las puntuaciones superiores a 0 representan un agotamiento significativo en la población mutante en comparación con la biblioteca de plásmidos original. Los genes se pueden dividir en tres grupos distintos que representan diferentes niveles de esencialidad en las líneas celulares. Esta agrupación en clústeres se puede utilizar para decidir el día de la ordenación. Si la pantalla se realiza en un punto de tiempo tardío (día 16), es posible que no se identifiquen genes que se sabe que son esenciales para las células (grupos 1 y 3). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla complementaria 1: Archivos de recuento sin procesar para y el software MAGeCK generado gene_summary archivos relacionados con las pantallas genéticas representativas. Haga clic aquí para ver este archivo (Haga clic con el botón derecho para descargar).
Tabla suplementaria 2: Agrupación de líneas celulares según la expresión de receptores de superficie celular. Haga clic aquí para ver este archivo (Haga clic con el botón derecho para descargar).
Discusión
Se describe una estrategia de cribado basada en CRISPR para identificar genes que codifican componentes celulares involucrados en el reconocimiento celular. Un enfoque similar mediante la activación de CRISPR también proporciona una alternativa genética para identificar los receptores que interactúan directamente con las proteínas recombinantes sin la necesidad de generar grandes bibliotecas de proteínas26. Sin embargo, una ventaja importante de este enfoque es que es aplicable a las interacciones mediadas por moléculas de superficie que se muestran de forma nativa en la célula y no depende de la sobreexpresión de los receptores, que puede influir en la avidez de unión del receptor. A diferencia de otros métodos, por lo tanto, esta técnica no hace suposiciones con respecto a la naturaleza bioquímica o la biología celular de los receptores y proporciona una oportunidad para estudiar interacciones mediadas por proteínas que normalmente son difíciles de estudiar utilizando enfoques bioquímicos, tales como proteínas muy grandes, o aquellos que atraviesan la membrana varias veces o forman complejos con otras proteínas, y moléculas distintas de proteínas como glicanos, glucólidos y fofolípidos. Dada la naturaleza a escala del genoma del método, este enfoque también tiene la ventaja de no sólo identificar el receptor, sino también los componentes celulares adicionales que se requieren para el evento de unión, proporcionando así información sobre la biología celular del receptor.
Una de las principales limitaciones de este método cuando se utiliza para identificar el receptor de una proteína huérfana es el requisito inicial de identificar primero una línea celular que se une a la proteína. Esto no siempre es fácil e identificar una línea celular que muestra un fenotipo de unión que también es permisivo para las pantallas genéticas puede ser el paso de limitación de tiempo para implementar este ensayo. Algunas líneas celulares tienden a unirse a más proteínas que otras. Esto es especialmente relevante para las proteínas que unen el HS, ya que estas proteínas tienden a unirse a cualquier línea celular que muestre las cadenas laterales del HS, independientemente del contexto de unión nativo. Además, hemos observado que la regulación ascendente de los sindecanos (es decir, los proteoglicaces que contienen ELC) en las líneas celulares conduce a una mayor unión de proteínas de unión al HS26. Esto podría ser un factor a tener en cuenta al seleccionar la línea celular para el cribado. Sin embargo, también es importante tener en cuenta que la unión aditiva del SE no interfiere con la unión a un receptor específico. Esto significa que si se observa la unión, es posible que esté mediada únicamente por el SE porque la unión mediada por el HS en este ensayo es aditiva y no codependiente19. En tal escenario, el enfoque de bloqueo de heparina descrito puede identificar tales comportamientos sin tener la necesidad de realizar una pantalla genética completa.
Un recurso útil para elegir líneas celulares es Cell Model Passport, que contiene información sobre genómica, transcriptómica y condición de cultivo para 1.000 líneas celulares de cáncer27. Dependiendo del contexto biológico, las células se pueden elegir en función de sus perfiles de expresión. Para ayudar a la selección de líneas celulares, agrupamos 1.000 líneas de celda en Cell Model Passport de acuerdo con la expresión de 1.500 glicoproteínas de superficie de células humanas preanotadas28 (Figura complementaria 2; información de clúster para cada línea de celda junto con las condiciones de crecimiento se proporcionan en la Tabla suplementaria 2). Al probar la unión de una proteína con función desconocida, es útil seleccionar un panel de líneas celulares representativas de cada clúster para aumentar la posibilidad de cubrir una amplia gama de receptores. Dada la elección, se recomienda elegir líneas celulares que sean fáciles de cultivar y fáciles de transducir. Dado que estas líneas celulares se utilizarán en el cribado a escala del genoma, es preferible que se puedan cultivar fácilmente en grandes cantidades y sean permisivas para la transducción lentiviral, ya que es el método más comúnmente disponible para la entrega de sgRNA para el cribado genético basado en CRISPR en los pasos posteriores.
Generalmente, las selecciones de fenotipo se llevan a cabo en una sola ordenación. Sin embargo, esto viene determinado por el brillo de la población de células manchadas en comparación con el control. Se podrían adoptar rondas iterativas de selecciones para escenarios en los que la relación señal-ruido del fenotipo deseado es baja, o cuando el objetivo de la pantalla es identificar mutantes que tienen fenotipos fuertes. Cuando se utiliza un enfoque de selección iterativa para pantallas basadas en FACS, es importante tener en cuenta que el proceso de clasificación puede causar la muerte celular, principalmente debido a la fuerza del clasificador. Por lo tanto, no todas las celdas recogidas se representarán en la siguiente ronda de clasificación.
La complejidad de la biblioteca es un factor muy importante para realizar pruebas genéticas exitosas, especialmente para las pantallas de selección negativas, ya que el grado de agotamiento en estos sólo se puede determinar comparando los resultados con lo que estaba presente en la biblioteca inicial. Para las pantallas de selección negativas, es común mantener bibliotecas de 500-1.000 x complejidad. Las pantallas de selección positivas, sin embargo, son más robustas para los tamaños de biblioteca, porque en tales pantallas sólo se espera que se seleccione un pequeño número de mutantes para un fenotipo en particular. Por lo tanto, en la pantalla de selección positiva descrita aquí, el tamaño de la biblioteca se puede reducir a 50-100x complejidad sin comprometer la calidad de la pantalla. Además, en estas pantallas también es posible utilizar una biblioteca de control para una línea celular determinada en un día determinado como un "control general" para todas las muestras ordenadas en el día para esa línea celular dada. Esto reducirá el número de bibliotecas de control que deben producirse y secuenciarse.
Otra consideración importante para el uso de este enfoque son las limitaciones de las pantallas de pérdida de función en la identificación de genes que son esenciales para el crecimiento celular in vitro. El momento de las pantallas es crucial en este sentido, ya que cuanto más tiempo se mantienen las células mutantes en el cultivo, mayor es la probabilidad de que las células con mutaciones en genes esenciales se vuelvan inviables y ya no estén representadas en la biblioteca mutante. Las recientes pruebas genéticas realizadas como parte de la iniciativa Project Score en más de 300 líneas celulares muestran que múltiples genes en la secreción de proteínas anotadas por KEGG y la vía de glicosilación N a menudo se identifican como esenciales para una serie de líneas celulares (Figura complementaria 3)29. Esto se puede tener en cuenta al diseñar pantallas si se debe investigar el efecto de los genes necesarios para la proliferación y la viabilidad en el contexto del proceso de reconocimiento celular. En este caso, la realización de pantallas en un punto de tiempo temprano (por ejemplo, el día 9 después de la traducción) sería generalmente apropiado. Sin embargo, si el enfoque se utiliza para identificar algunos objetivos con efectos de tamaño fuerte en lugar de vías celulares generales, podría ser apropiado realizar pantallas en un momento posterior (por ejemplo, día 15-16 después de latransducción).
Los resultados del cribado son muy robustos; en ocho pantallas de unión a proteínas recombinantes realizadas en el pasado, el receptor de superficie celular fue el principal golpeado en cada caso19. Al utilizar este enfoque para identificar al socio de interacción, por lo tanto, uno debe esperar que el receptor y los factores que contribuyen a su presentación en la superficie se identifiquen con una alta confianza estadística. Una vez que se realiza la pantalla y se valida un golpe utilizando un solo knockout de gRNA, se pueden realizar seguimientos adicionales utilizando métodos bioquímicos existentes como AVEXIS4 y la unión saturable directa de proteínas purificadas utilizando resonancia de plasmon superficial. El enfoque descrito aquí es aplicable a todas las proteínas para las que es posible generar una sonda de unión recombinante soluble.
En resumen, se trata de un enfoque CRISPR knockout a escala del genoma para identificar interacciones mediadas por proteínas de superficie celular. Este método es generalmente aplicable para identificar las vías celulares necesarias para el reconocimiento de la superficie celular en una amplia gama de contextos biológicos diferentes, incluso entre las propias células de un organismo (por ejemplo, reconocimiento neuronal e inmunológico), así como entre las células huésped y las proteínas patógenas. Este método proporciona una alternativa genética a los enfoques bioquímicos diseñados para la identificación de receptores, y debido a que no requiere ninguna suposición previa con respecto a la naturaleza bioquímica o biología celular de los receptores tiene un gran potencial para hacer descubrimientos completamente inesperados.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue apoyado por la subvención Wellcome Trust número 206194 otorgada a GJW. Agradecemos a las instalaciones de Cytometry Core: Bee Ling Ng, Jennifer Graham, Sam Thompson y Christopher Hall por ayuda con FACS.
Materiales
| Name | Company | Catalog Number | Comments |
| Anti-mouse alkaline phosphatase | Sigma | A4656 | |
| Blasticidin | Chem-Cruz | SC-204655 | |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | 13362 | |
| BSA | Sigma | A9647-100G | |
| Diethanolamine | Sigma | 398179 | |
| DMEM | Gibco | 31966-021 | |
| Dneasy Blood and Tissue kit | Qiagen | 69504 | |
| DynaMag-96 Side Magnet | Invitrogen | 12331D | |
| HEK293T packaging cells | ATCC | CRL-3216 | |
| Heparin | Sigma | H4784-1G | |
| KAPA HiFi HotStart ReadyMix | Kapa | KK2602 | |
| Lipofectamine LTX with PLUS reagent | Invitrogen | 15338100 | |
| MoFlo XDP cell sorter | BD | ||
| Ni2+-NTA agarose beads | Jena Bioscience | AC-501-25 | |
| OPTI-MEM | Life Technologies | 31985-070 | |
| OX-68 antibody | AbD Serotec | MCA1022R | |
| p-nitrophenyl phosphate | Sigma | 1040-506 | |
| PD-10 desalting columns | GE healthcare | 17085101 | |
| Polybrene | Millipore | TR-1003-G | |
| Polypropylene tubes with 5 mL bed volume | Qiagen | 34964 | |
| Proteinase K, recombinant, PCR Grade | Roche | 3115879001 | |
| Puromycin | Gibco | A11138-03 | |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | M0494L | |
| QIAquick PCR purification kit | Qiagen | 28104 | |
| SCFA filter | Nalgene | 190-2545 | |
| Sony Cell sorter | Sony | ||
| SPRI beads (Agencourt AMPure XP beads) | Beckman | A63881 | |
| Streptavidin-coated microtitre plates | Nalgene | 734-1284 | |
| Streptavidin-PE | Biolegend | 405204 |
Referencias
- Wright, G. J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems. 5 (12), 1405-1412 (2009).
- van der Merwe, P. A., Barclay, A. N. Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends in Biochemical Sciences. 19 (9), 354-358 (1994).
- Wood, L., Wright, G. J. Approaches to identify extracellular receptor-ligand interactions. Current Opinion in Structural Biology. 56, 28-36 (2019).
- Bushell, K. M., Söllner, C., Schuster-Boeckler, B., Bateman, A., Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Research. 18 (4), 622-630 (2008).
- Visser, J. J., et al. An extracellular biochemical screen reveals that FLRTs and Unc5s mediate neuronal subtype recognition in the retina. eLife. 4, e08149 (2015).
- Özkan, E., et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell. 154 (1), 228-239 (2013).
- Martinez-Martin, N., et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell. 174 (5), 1158-1171 (2018).
- Bianchi, E., Doe, B., Goulding, D., Wright, G. J. Juno is the egg Izumo receptor and is essential for mammalian fertilization. Nature. 508 (7497), 483-487 (2014).
- Mullican, S. E., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature Medicine. 23 (10), 1150-1157 (2017).
- Turner, L., et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 498 (7455), 502-505 (2013).
- Frei, A. P., et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nature Biotechnology. 30 (10), 997-1001 (2012).
- Sobotzki, N., et al. HATRIC-based identification of receptors for orphan ligands. Nature Communications. 9 (1), 1519 (2018).
- Sharma, S., Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 19 (4), (2018).
- Gebre, M., Nomburg, J. L., Gewurz, B. E. CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses. 10 (2), (2018).
- Zotova, A., Zotov, I., Filatov, A., Mazurov, D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. Journal of Immunological Methods. 439, 8-14 (2016).
- Puschnik, A. S., Majzoub, K., Ooi, Y. S., Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nature Reviews. Microbiology. 15 (6), 351-364 (2017).
- Kerr, J. S., Wright, G. J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. Journal of Visualized Experiments. (61), e3881 (2012).
- Tzelepis, K., et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Reports. 17 (4), 1193-1205 (2016).
- Sharma, S., Bartholdson, S. J., Couch, A. C. M., Yusa, K., Wright, G. J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Research. 28 (9), 1372-1382 (2018).
- Wang, B., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nature Protocols. 14 (3), 756-780 (2019).
- Hart, T., et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3. 7 (8), 2719-2727 (2017).
- Kuleshov, M. V., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44 (W1), W90-W97 (2016).
- Crosnier, C., et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 480 (7378), 534-537 (2011).
- Kirk, P., et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal. 19 (15), 3896-3904 (2000).
- Chong, Z. S., Ohnishi, S., Yusa, K., Wright, G. J. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biology. 19 (1), 205 (2018).
- van der Meer, D., et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Research. 47 (D1), D923-D929 (2019).
- Bausch-Fluck, D., et al. A mass spectrometric-derived cell surface protein atlas. PloS One. 10 (3), e0121314 (2015).
- Behan, F. M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 568 (7753), 511-516 (2019).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados