
Method Article
ゲノムスケールCRISPR/Cas9遺伝子スクリーンを用いた細胞表面受容体同定
要約
本稿では、細胞外受容体とリガンドの相互作用を同定するためのゲノムスケール細胞ベースのスクリーニングアプローチについて述べる。
要約
膜埋め込み細胞表面受容体間の直接的な相互作用によって媒介される細胞間通信は、多細胞生物の正常な発達と機能にとって極めて重要である。しかし、これらの相互作用を検出することは技術的に困難です。本稿では、特異的細胞表面認識事象に必要な細胞経路を明らかにする、体系的なゲノムスケールCRISPR/Cas9ノックアウト遺伝子スクリーニングアプローチについて述べる。このアッセイは、哺乳動物タンパク質発現系で産生される組換えタンパク質を、細胞ベースの遺伝的スクリーンにおける相互作用パートナーを同定するための熱心な結合プローブとして利用する。この方法は、膜組み込み受容体のエクトドメインに対応する組換え結合プローブによって検出される細胞表面相互作用に必要な遺伝子を同定するために使用することができる。重要なことに、このアプローチのゲノムスケールの性質を考えると、直接受容体を同定するだけでなく、細胞表面での受容体の提示に必要な細胞成分を同定する利点もあり、それによって受容体の生物学に関する貴重な洞察を提供する。
概要
細胞表面受容体タンパク質による細胞外相互作用は、組織組織、宿主-病原体認識、免疫調節などの重要な生物学的プロセスを直接的に行う。膜受容体はモノクローナル抗体などの体系的に送達された治療薬の有効な標的であるため、これらの相互作用を調査することは、より広範な生物医学界に興味深いものである。その重要性にもかかわらず、これらの相互作用を研究することは技術的に困難なままです。これは主に、膜組み込み受容体が両生性であり、生化学的操作のために生物学的膜から分離することが困難であり、それらの相互作用は弱い相互作用親和性(μM-mM範囲のKKDs)1に代表される。1その結果、多くの一般的に使用される方法は、タンパク質相互作用のこのクラスを検出するのに適していません1,,2.
細胞外受容体リガンド相互作用を考慮に入れた細胞外受容体リガンド相互作用を考慮して、さまざまな方法が開発されている。これらのアプローチの多くは、哺乳類または昆虫細胞系で可溶性組換えタンパク質として受容体の全ての異性ドメインを発現することを含み、これらのタンパク質がグリカンおよびジスルフィド結合などの構造的に重要な翻訳後修飾を確実に含んでいる。低親和性結合を克服するために、エクトドメインは、しばしばそれらの結合性を増加させるためにオリゴマー化される。アビッドタンパク質のエクトドメインは、直接組換えタンパク質-タンパク質相互作用スクリーン4、5、6、75,6における4相互作用パートナーを同定するための結合プローブとして正常に使用されている。,7広く成功しているが、組換えタンパク質ベースの方法は、膜受容体のエクトドメインが可溶性タンパク質として産生されることを必要とする。したがって、連続した細胞外領域(例えば、シングルパスタイプI、II型、またはGPIアンカー)を含むタンパク質に対してのみ一般的に適用可能であり、膜に複数回及ぶ受容体複合体および膜タンパク質には一般的には適していません。
相補的なDNA(cDNA)のライブラリーを細胞にトランスフェクトし、結合性表現型の利益について試験した発現クローニング技術も、細胞外タンパク質とタンパク質相互作用を同定するために使用されている。近年、クローン化および配列配列されたcDNA発現プラスミドの大規模なコレクションの入手可能性は、細胞表面受容体をコードするcDNAを過剰発現する細胞株をスクリーニングし9、相互作用を同定するために組み換えタンパク質の結合をスクリーニングする方法を促進している。cDNA過剰発現ベースのアプローチは、組換えタンパク質ベースの方法とは異なり、原形質膜の文脈における相互作用を同定する可能性を与える。しかし、cDNA発現構築物を使用する成功は、正しく折り畳まれた形でタンパク質を過剰発現させる細胞の能力に依存しますが、これはしばしばトランスポーター、シャペロン、および正しいオリゴマーアセンブリなどの細胞アクセサリー因子を必要とします。したがって、単一のcDNAをトランスフェクトするだけでは、細胞表面の発現を達成できない場合があります。
cDNA構築物または組換えタンパク質プローブを使用したスクリーニング技術は、リソースを大量に消費し、大量のcDNAまたは組換えタンパク質ライブラリーを必要とします。最近では、大規模なライブラリの組み立てを必要としない細胞外相互作用を同定するために、質量分析法を用いた具体的な設計法が利用されている。しかしながら、これらの技術は細胞表面の化学的操作を必要とし、細胞表面に存在する分子の生化学的性質を変化させることができ、現在はグリコシル化タンパク質11,12,12によって媒介される相互作用にのみ適用可能である。現在入手可能な方法の大半は、グリカン、脂質、コレステロールなどの分子を含む膜微小環境からの寄与をほとんど無視しながら、タンパク質間の相互作用に重点を置いています。
CRISPRベースのアプローチを用いた高効率のバイアレリックターゲティングの最近の開発により、定義された遺伝子を欠く細胞のゲノムスケールライブラリは、異なる文脈に関与する細胞成分を同定するための体系的かつ公平な方法でスクリーニングすることができる単一のプールで、 細胞シグナル伝達プロセスの解剖、薬物、毒素、病原体に対する耐性を付与する摂動の同定、および抗体13、14、15、16,15,の特異性の決定を含む。13,16ここでは、細胞外受容体とリガンド相互作用を同定するための現在の生化学的アプローチに代わるゲノムスケールCRISPRベースのノックアウト細胞スクリーニングアッセイについて述べている。遺伝子スクリーンによって膜受容体によって媒介される相互作用を同定するこのアプローチは、cDNAまたは組換えタンパク質の大規模なライブラリをコンパイルする必要を回避するため、個々のリガンドに焦点を当てた研究者に特に適しています。
このアッセイは、3つの主要なステップからなる:1)対象の受容体の細胞外領域からなる非常にアヴィッドな組換えタンパク質結合プローブが生成され、蛍光ベースのフローサイトメトリーベースの結合アッセイで使用される。2)結合アッセイは、組換えタンパク質プローブの相互作用パートナーを発現する細胞株を同定するために使用される。3) 目的のタンパク質と相互作用する細胞株のCas9発現バージョンが生成され、ゲノムスケールCRISPR/Cas9ベースのノックアウト画面が行われる(図1)。この遺伝子スクリーンでは、組換えタンパク質と細胞株への結合は、プローブに結合する能力を失ったノックアウトライブラリー内の細胞を、蛍光ベースの活性化細胞分類(FACS)およびシーケンシングによって同定された結合表現型の喪失を引き起こした遺伝子を用いて分類する測定可能な表現型として使用される。原則として、熱心なプローブと細胞表面表示に必要な結合を担う受容体をコードする遺伝子が同定される。
このプロトコルの最初のステップは、膜結合受容体のエクトドメインを表す熱心な組換えタンパク質プローブの産生を含む。これらの受容体は、それらのエクトドメインが組換え可溶性タンパク質1として発現する場合、しばしば細胞外結合機能を保持することが知られている。目的のタンパク質については、可溶性組換えタンパク質は、結合性を高めるためにオリゴマー化することができる場合であれば、任意の形式で適切な真核生物タンパク質発現系で産生することができ、蛍光ベースのフローサイトメトリーベースの結合アッセイ(例えば、FLAGタグ、ビオチンタグ)に使用できるタグが含まれています。HEK293タンパク質発現系を用いた膜受容体の可溶性異物ドメインの製造に関する詳細なプロトコル、ならびにペンタマータンパク質および単量体タンパク質の両方の産生のための異なる多量体化技術およびタンパク質発現構築物については、前に1,17,17を説明した。ここでのプロトコルは、単量体のビオチン化されたタンパク質からフルオロクロム(例えば、フィコエリスリン、またはPE)に結合させることによって蛍光性有数のプローブを生成するステップを説明する。ゲノムスケールスクリーンを行うための一般的なプロトコルは、すでに20,21,21に記載されており、したがって、プロトコルは、主にヒトV1(「ゆす」)ライブラリ18を用いたCRISPR/Cas9ノックアウトスクリーニングシステムを用いてフローサイトメトリーベースの組換えタンパク結合スクリーンを行う詳細に焦点を当てている。
プロトコル
1. ビオチン化されたHisタグ付きタンパク質の製造と精製
- 哺乳類または昆虫細胞ベースのタンパク質発現系を用いて、可溶性組換えのHisタグ付きビオチン化タンパク質を生成する(表1のプラスミド構築物を参照)。
注:HEK293細胞発現系を用いた単量体ビオチンおよびHisタグ付きタンパク質の製造に関する詳細なプロトコルは、Kerr etal.HEK293発現系を用いて発現するタンパク質エクトドメインは、培養培地中に分泌される。 - 3,000 x gで 20 分間遠心分離して細胞をペレット化することにより、可溶性タンパク質を収集します。
- 上清を0.22μmのフィルターでフィルターし、1:1,000比で濾過したタンパク質上清にNi 2+-NTAアガロースビーズを加えます(すなわち、50%のアガローススラリーの50μLを50mLの上清に加えます)。回転プラットフォーム上で4°Cで一晩または少なくとも4〜5時間インキュベートする。
- ポリプロピレンカラムを5 mLのHis精製洗浄バッファーを加えて洗浄します。すべてのバッファー構成については、表 2を参照してください。
- ビーズとタンパク質の上清混合物全体をカラムに注ぎます。ビーズはベースに蓄積されます。
- 15 mLの洗浄バッファーでビーズ2倍を洗います。タンパク質希釈を避けるために、5 mLのシリンジでカラムから残った洗浄バッファーを慎重に引き出し、廃棄します。
- 300~500μLのHis精製溶出バッファーをビーズに直接加え、少なくとも1時間インキュベートします。脱塩カラムを用いて、溶出バッファーを所望の緩衝液(例えば、通常はPBSまたはHBS)に交換する。すべてのタンパク質を4°Cで保存し、さらに使用するまで保管してください。
2. 単量体ビオチン化タンパク質の定量とオリゴマー化
注:結合の結合の熱心さを高めるために、結合アッセイでそれらを使用する前に四量体連鎖球状のストレプトアビジン-PE上のビオチン化単量体タンパク質をオリゴマー化する。一定濃度のストレプトアビジンに対して一連のビオチン化モノマーを試験し、過剰なビオチン化モノマーを検出できない最小希釈を実証的に確立することにより、単量体タンパク質および四量体ストレプトアビジン-PEの最適な結合比を達成する。
- 96ウェルプレートに適切な希釈バッファー(1%ウシ血清アルブミン[BSA]を有するPBSまたはHBS)を使用して、ビオチン化タンパク質サンプルの少なくとも8つの連続希釈液を作ります。各希釈液の最終容積が少なくとも200 μLであることを確認してください。
- 各ウェルから100 μLを取り出し、新しい96ウェルプレートに移して、サンプルのプレートを複製します。常にコントロールを含めます。この場合、コントロールはタグのみのタンパク質(すなわち、ビオチン化Hisタグ付きCd4ドメイン3+4タンパク質)である。これは、すべての結合アッセイにおいて制御プローブとして使用される。
- 希釈バッファーでストレプトアビジン-PEを 0.1 μg/mL に希釈します。
- プレートの1つだけに、希釈されたストレプトアビジン-PEの100 μLを加えます。重複プレートはコントロールとして機能します。制御プレートに100 μLの希釈バッファーを加えて、ボリュームを均等にします。
- 室温(RT)で20分間インキュベートします。その間、ストレプトアビジンコーティングされたプレートのウェルを希釈バッファーで15分間ブロックします。
- 両方のプレートからサンプルの総体積をストレプトアビジンコーティングされたプレートの個々のウェルに移し、RTで1時間インキュベートします。
- プレート3xを200 μLの洗浄バッファー(すなわち、0.1%Tween-20,2%BSAでPBSまたはHBS)で洗浄します。2 μg/mL マウスアンチラット Cd4d3+4 IgG (OX68) の 100 μL を加え、RT で 1 時間インキュベートします。
- 洗浄バッファーでプレート3xを洗浄します。RTで1時間0.2 μg/mLで抗マウスアルカリホスファターゼコンジュゲートの100 μLを加えます。
- 洗浄バッファーでプレート3xを洗浄し、希釈バッファーで1xを洗います。
- ジエタノールアミンバッファーで 1 mg/mL で p-ニトロフェニル リン酸を準備します。.各ウェルに100 μLを加え、15分間インキュベートします。
- 405 nmで吸光度の測定値を取ります。プレート上に信号がない最小希釈を適切な希釈係数として使用して四倍数子を作成します (図 2)。
- 4 μg/mLストレプトアビジン-PEと適切なビオチン化タンパク質希釈をRTで30分間インキュベートすることで、すべてのサンプルとコントロールに対して10倍の四量体染色液を作り、さらに使用するまで4°Cの光保護チューブにコンジュゲートタンパク質を保存します。
3. フローサイトメトリーベースの細胞結合アッセイ
- 接着細胞の場合は、培養培地を除去し、2価カチオンなしでPBSで1xを洗浄します。次に、細胞剥離液(例えば、EDTA)を追加する。細胞を5〜10分間取り外し、フラスコを軽くたたいて細胞を放出します。
注意: トリプシンベースの製品は細胞表面タンパク質を切断できるため、使用しないでください。 - チューブに切り離された細胞を収集します。懸濁液中に増殖する細胞(例えば、HEK293細胞)については、培養フラスコから直接細胞をチューブに集める。
- ペレット細胞は200 x gで5分間、上清を取り除き、洗浄バッファー(すなわち、PBS/1%BSA)でペレットを再懸濁します。
- ヘモサイトメーターを使用して細胞を数え、濃度を2.5 x 105-1 x 106細胞/mLに調整します。アリコート 100 μL の調製セルミックスを 96 ウェル U-または V- 底板に取り付けました。400 x g で 5 分間プレートを回転させます。マルチチャンネルピペットで上清を取り外します。
- 正規化された蛍光標識された高いアビッドタンパク質プローブおよびコントロールを細胞で以前に調製したプレートに100 μL加え、4°Cで1時間インキュベートします。1時間の結合後、プレートを400xgでg5分間回転させます。
- 上清を取り出し、200 μLの洗浄バッファー(すなわち、PBS/1% BSA)を加えます。上下にピペットを入れ、よく混ぜます。
- ペレット細胞を400xgで5分間遠心分離してg、洗浄工程1倍を繰り返します。2回のスミを行った後、上清を完全に取り除き、PBSの100 μLで細胞ペレットを再懸濁します。
- フローサイトメトリーで細胞を分析します。黄緑色のレーザー(すなわち、561 nm)を使用して、PE蛍光を検出します。
- まず、コントロールプローブで染色された細胞を分析します。PE蛍光の分布に基づいて、このゲートに1%以下のコントロール細胞が落ちるような結合集団のためのゲートを描く。
- サンプルを分析し、結合ゲートに入るセルの割合を特定します。
注: より高い結合集団を表示する細胞株は、より高いシグナル対雑音比を有するため、遺伝的スクリーンに望ましい。理想的には、細胞の80%以上がこのゲート内に入るはずです。
4. 熱不安定エピトープおよびヘパラン硫酸サイドチェーンからの結合寄与の決定
注:多くのタンパク質の活性は熱不安定であるため、熱処理後の結合活性の喪失は励みになります。組換えタンパク質の結合を媒介する際の負の電荷グリコサミノグリカン(主にヘパラン硫酸塩(HS))からの寄与を決定することをお勧めします。これは、ここで説明する細胞結合アッセイにおけるHSによる結合は、他の受容体19に共依存するのではなく、添加剤であり得るからである。これは、観察された結合は、特異的受容体ではなく、細胞表面プロテオグリカンのHS側鎖によって完全に媒介され得る。細胞表面上のHSへの結合は必ずしも非特異的ではなく、むしろタンパク質の特性であり、完全な遺伝的スクリーンを実行する前に知っておくのが有用である。
- 結合アッセイで使用する熱処理タンパク質サンプルを準備します。
- 正規化されたが共役していない単量体タンパク質を80°Cで10分間加熱します。
- 熱処理されたタンパク質を、ELISAで決定した未処理のタンパク質と同じ結合比を仮定してストレプトアビジン-PEに結合する(セクション2参照)。
- ヘパリンブロックタンパク質サンプルを準備します。
- 2 mg/mL の開始濃度と 100 μL の最終容積で PBS で 8 つの 1:3 希釈液を PBS で調製します。
- ヘパリン希釈液中に100μLの調製された結合プローブを少なくとも30分間インキュベートします。
- 第3項に記載の結合アッセイで、熱処理タンパク質およびヘパリン/タンパク質混合物の完全な200 μLを使用する。代表的な結果は、図3A,Bに示されています。
5. Cas9を安定的に表現する細胞株の選択
注:目的のプローブを結合する細胞株をCRISPRスクリーニングで使用する前に、Cas9ヌクレアーゼと高度に活性なクローンを19に選択して発現するように設計する必要があります。
- 以下の一般的なレンチウイルス産生プロトコルを使用して、Cas9発現用レンチウイルス構造を用いてレンチウイルスを産生する(表1参照)。
- 培養 HEK293-FT細胞をDMEM/10% FBS培地中で37°Cおよび5%CO2で培養する。種子HEK293-FT細胞はトランスフェクションの1日前にトランスフェクションの日に約80%コンフルエントになるようにします。
注:HEK293FT細胞は緩やかに接着性があります。したがって、レンチウイルスの製造に使用する場合は、0.1%(w/v)ゼラチンをコーティングした培養フラスコにめっきして付着性を高めてみてください。 - 午前中にトランスフェクションを行います。トランスファーベクター、パッケージミックス、トランスフェクション試薬を、前温めトランスフェクション適合媒体(例えばOpti-MEM)に添加します。チューブ10-15倍を反転して混合します。RTで5分間インキュベートを行う。 Table 3
- メーカーが提案したトランスフェクション試薬を追加します。クイック渦によって混ぜます。RTで30分間インキュベートします。
- 非常に慎重に使用済み培地を吸引します。プレートにトランスフェクション対応のメディアを追加します。
- トランスフェクション試薬/DNA複合体をプレートの側面に滴下し、非常に穏やかに渦巻くことによってゆっくりとプレートを通して広がります。
- 37°Cで3~5時間インキュベートし、培地をD10培地に交換します。一晩インキュベート。
- 翌日の朝、新鮮なD10培地に交換してください。一晩インキュベート。
- 翌日の午後遅く、ウイルス上清を集める。0.45 μm のフィルターで、低タンパク質結合性を持つフィルター。必要に応じて、新鮮なD10培地を追加し、一晩インキュベートし、翌日上清を回収します。
- ウイルス上清は、4°Cで数日間だけ安定している。長期保存のため-80°Cで保管してください。
注:細胞をトランスデューシングするのが困難な伝達のために望ましいと考えられる高濃度レンチウイルス製剤を生成するために、上清はまた、4°Cで一晩6,000 x gで遠心分離によって濃縮することができる。半透明のウイルスペレットにエタノール耐性ペンでマークし、上清を捨てます。元の体積の1/100分の1にペレットを再懸濁し、濃度を100倍に増やします。
- 培養 HEK293-FT細胞をDMEM/10% FBS培地中で37°Cおよび5%CO2で培養する。種子HEK293-FT細胞はトランスフェクションの1日前にトランスフェクションの日に約80%コンフルエントになるようにします。
- レンチウイルスで細胞をトランスデュースする。
- 3 mLの適切な培養培地を用いた6ウェルプレートに1ウェルあたり1 x106細胞をプレートする。一部の細胞は、他の細胞よりも容易に伝達されます。細胞(例えばHEK細胞)をトランスデュースしやすい場合は、レンチウイルスを細胞に直接加える。細胞をトランスデュースすることが困難な場合、以下に説明するように、スパイクーのプロトコルに従う必要があるかもしれません。
- アリコート2 mLの2-5 x 106細胞/mLを15 mL円錐管に入した。
- レンチウイルスを8μg/mLヘキサジメトリン臭化物と一緒に加え、RTで30分間インキュベートします。
- 遠心分離機は32°Cで800×ggで100分間次に、同じ培地中の細胞を再懸濁し、適切な培地を用いて適切な培養フラスコに細胞懸濁液を加える。
- 少なくとも24時間の伝達を許可します. その後、ウイルスを含むメディアを削除し、新鮮な媒体を追加します。
- 24時間後、適切な抗生物質を補充したものに培地を変更する。Cas9構造は選択のためのブラストシジン抵抗カセットを含んでいる。
注: ブラストシジジンの量は、用量応答キルカーブを実行することにより、各細胞株に対して最適化する必要があります。2.5~50 μg/mLの間のブラストシジン濃度は、トランスダクションの10日以内に、ほとんどの未透過細胞株を殺す必要があります。
- 3 mLの適切な培養培地を用いた6ウェルプレートに1ウェルあたり1 x106細胞をプレートする。一部の細胞は、他の細胞よりも容易に伝達されます。細胞(例えばHEK細胞)をトランスデュースしやすい場合は、レンチウイルスを細胞に直接加える。細胞をトランスデュースすることが困難な場合、以下に説明するように、スパイクーのプロトコルに従う必要があるかもしれません。
- コントロールプレート内のすべての細胞(すなわち、同じ濃度の選択抗生物質で処理された非透過細胞)が殺されるまで選択を行う。
6. 高Cas9-アクティビティクローンの選択
注:ポリクローナルCas9は、正常に遺伝的スクリーンを実行するために使用することができます。しかし、高いCas9活性を有するクローンを選択すると、スクリーニング結果18が向上する。
- 限定希釈または単一細胞ソート個々のブラストシジン耐性細胞を、ブラストシジンを補充した培養培地を含む3つの96ウェルプレートのウェルに分類する。クローンは2-4週間の間に出現し始めます。10~20クローンを選択し、6ウェルプレートに展開します。
- Cas9活性のクローンを、gRNA標的GFPまたは空のgRNAを制御18として発現する構築物を用いて細胞をトランスサイジングする外因性遺伝子ノックアウトシステムを使用してCas9活性のクローンをアッセイする。
- 注文レポータープラスミド:GFP-BFPプラスミド、コントロールBFPプラスミド(表1)
- セクション5.1で説明されているレンチウイルス産生プロトコルを使用して、GFP-BFPプラスミドおよびコントロールBFPプラスミドの両方にレンチウイルスを産生する。
- GFP-BFPシステムとControl-BFPをコードするレンチウイルスを用いて、Cas9発現細胞株クローンをそれぞれ個別にトランスデューシングします。セクション 5.2 のプロトコルに従ってください。
- 3日間の導入後、フローサイトメトリーを用いて各クローンのGFP-BFP蛍光を調べた。488 nm レーザーと 405 nm レーザーを使用して、それぞれ GFP と BFP を検出します。
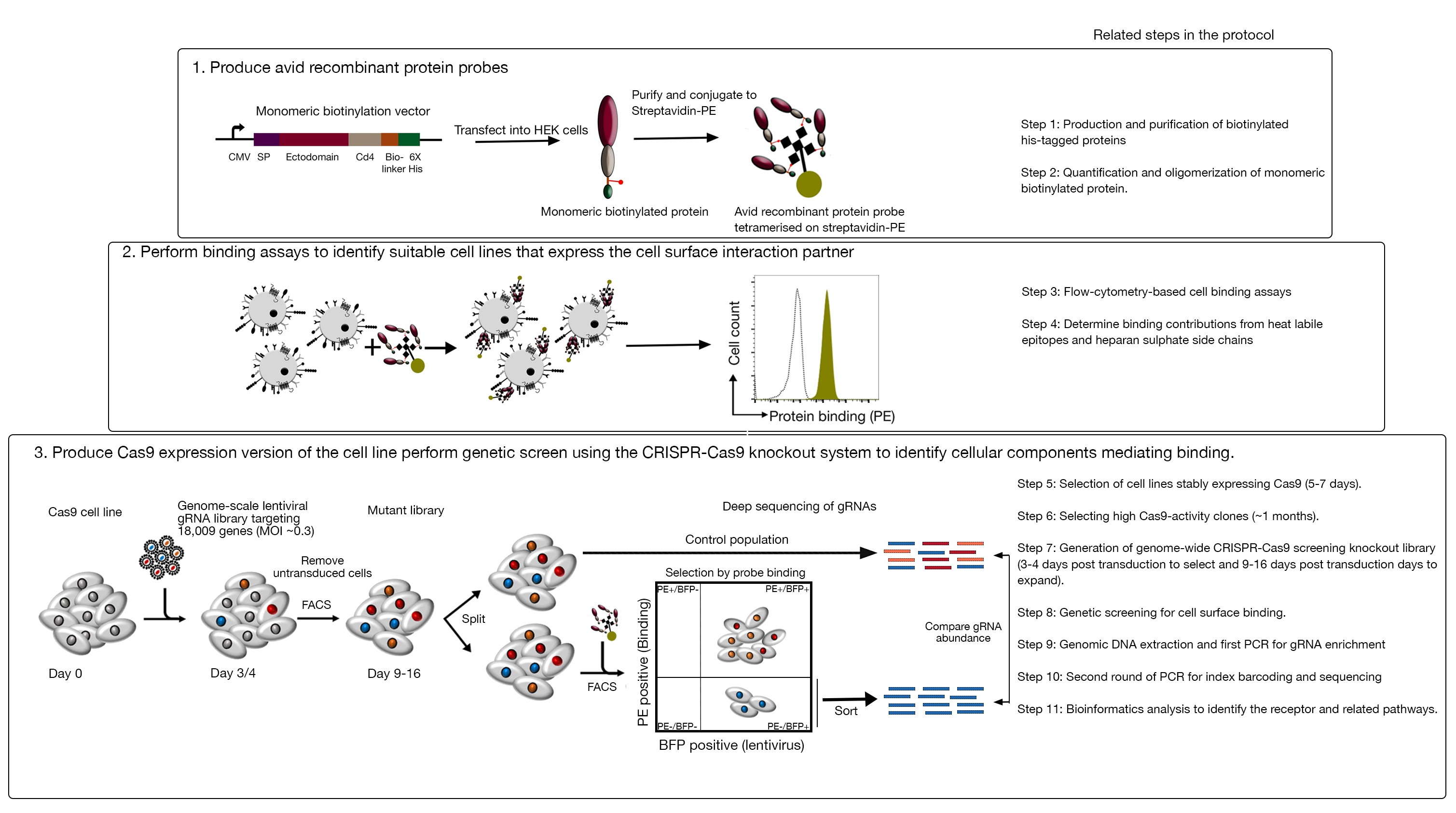
- GFP-BFP-二重陽性細胞に対するBFPの比率のみを調べることにより、各クローンのCas9活性を定量化します。高活性 Cas9 細胞は、理想的には、>95% GFP ノックアウト効率を持つ必要があります (図 4)。
7. ゲノムワイドCRISPR-Cas9スクリーニングノックアウトライブラリーの生成
- Human V1ライブラリ18を用いたゲノムワイドスクリーニングについては、ゲノム全体のライブラリを注文し(表1参照)、製造者マニュアルの「ライブラリ複製プロトコル」に記載されているプロトコルを使用して細菌刺しからプラスミドライブラリを調製します。
- ゲノムワイドライブラリプラスミド製剤を使用して、セクション5.1に記載されているレンチウイルス産生プロトコルを使用してヒト遺伝子の標的破壊のためのgRNAをコードするレンチウイルスライブラリーを作製する。
注:良い方法は、実験的な一貫性を向上させるために、トランスダクション用に最適化されたレンチウイルス製剤の単一のバッチを生成することです。 - セクション5.2のトランスダクションプロトコルを使用して、小規模なテストトランスダクションを行い、30%のトランスダクションを達成するために各細胞株に必要なウイルス量を決定します。フローサイトメトリーを使用して、BFP蛍光をトランスダクション効率の代理として評価します。
- HEK293細胞をトランスデュースするには、所定のレンチウイルス製剤を、〜4時間の正常増殖培地で培養した30〜50 x106細胞に添加する。その後、レンチウイルスでメディアを取り出し、新鮮な成長培地に置き換えます。
- 他の細胞株の場合は、セクション5.2.1のスパイク法を使用しますが、より大きなスケールで、合計30〜50 x 106個の細胞が変換されるようにします。このために、アリコート2mLの5 x 106細胞/mLを15mL円錐管で、指示に従って進行する。
- 接着性細胞株については、トランスダクション後24時間にピューロマイシンを添加して細胞をトランスデューシングした細胞を選択する。
注:用量応答キルカーブを実行することにより、ピューロマイシン濃度を最適化します。通常、1~10 μg/mLの間の濃度は、3-5日以内に非透過細胞を殺す必要があります。これは、複数の単一のガイドRNA(sgRNA)によって導入された細胞を選択する可能性を高める可能性があるため、より高濃度のピューロマイシンを使用しないでください。 - 懸濁細胞の場合、細胞ソーターを用いて3日後に細胞を導入した(すなわちBFP陽性)細胞を収穫し、少なくとも10 x106細胞を含むライブラリーを生成する。BFPを用いて選択したら、適切な量のピューロマイシンを補充した培地中の細胞を増殖させる。
注:細胞の選別を妨げる可能性のある細胞培養物から死んだ細胞や残骸を除去することは困難であるため、懸濁細胞株のみのピューロマイシンでの選択は避けてください。 - 培養変異体ライブラリーは、2〜3日ごとに規則的な通路を伴う9〜16日間の転帰後に用いた。
8. 細胞表面結合の遺伝子スクリーニング
- 200 x gで変異細胞ライブラリーをペレット化し、5分間、PBS中の細胞を再懸濁させた。
- 各チューブに少なくとも50 x 106個の細胞を有する2つの15 mL円錐管に細胞を分割します。
- 200 x gで 1 本の円錐チューブを 5 分間回転させ、上清を取り除き、細胞ペレットを -20 °C で凍結します。これはコントロールの母集団であり、後で処理されます。
- 他のチューブのペレットをPBS/1%BSAの10 mLに再懸濁します。96ウェルプレートに負のコントロールとして細胞の100 μLを確保します。
- 適切な組合体組換えタンパク質を円錐チューブの細胞懸濁液に加え、陰性対照タンパク質を96ウェルプレートに加えます。
- 緩やかな回転(6 rpm)のベンチトップローターで4°Cで少なくとも1時間の細胞染色を行う。
- 細胞を200xgで5g分間ペレットし、上清を取り除く。2回の洗浄工程を行い、その後、細胞を5mLのPBSで再懸濁する。
- 30 μmの細胞ストレーナーを使用して細胞を歪み、細胞クラスタを除去します。フローソーターを使用して分析します。
- BFP+/PE-セルのゲートには、陰性コントロールサンプルを使用します。
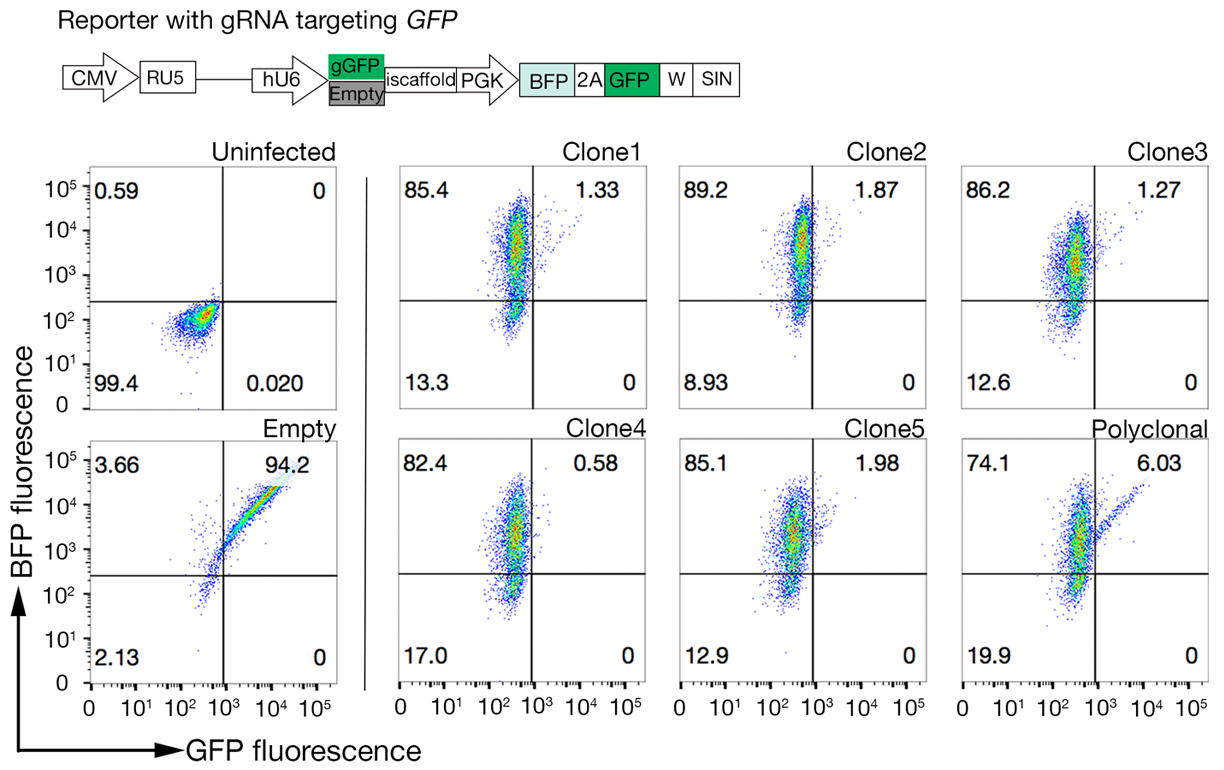
- サンプルをソートし、BFP+/PE細胞を収集します。並べ替えゲートは、タンパク質への細胞の結合に依存しますが、通常PE陰性サンプルの1〜5%が収集されます。ソート ゲートの例は補助図 1に示されています。
- 選択したゲートから 500,000 ~1,000,000 個のセルを収集します。細胞数が少ない場合、損失を最小限に抑えるために、1.5 mL遠心チューブにサンプルを採取することを検討してください。
- 500 x gで 5 分間遠心分離して選別した細胞をペレットにします。ペレットを-20°Cで6ヶ月間保存することが可能です。
9. gRNA濃縮のためのゲノムDNA抽出および最初のPCR
- 高複雑性制御集団からゲノムDNAを抽出する。
- 対照集団が-20°Cで凍結した場合は、円錐管を取り出し、PBSを加える。氷の上に置いてペレットを解凍してください。
- 50 x 10 6細胞からゲノムDNAを抽出するために製造業者の推奨を使用して、市販キット(材料表を参照)を使用してください。DNA濃度を1mg/mLに調整してください。
- サンプルごとに、72 μgのDNAに対応するPCR用のマスターミックスを設定します。アリコートは、96ウェルPCRプレートの36ウェルでウェルあたり50 μL。必要なプライマーシーケンスを表4に示します。表 5および表6のガイドを使用します。
- 2%(w/v)アガロースゲルで6-12代表サンプルからPCRの5μLを解決します。〜250 bpで単一のクリアバンドが観察されるべきである。バンドが薄い場合は、PCR を 2 ~ 3 サイクル繰り返します。
- マルチチャネルピペットを使用して、各ウェルから5 μLのPCR製品(合計180 μL)を収集し、商業キットから900 μLの結合バッファーを含むリザーバにプールします(材料表を参照)。
- 市販の PCR 精製キットを使用して PCR 製品を精製します。市販キットから50μLの溶出バッファーにDNAを溶出し(材料表を参照)、DNA濃度を測定します。
- 結合表現型の損失のためにソートされたサンプルは、多数の独立クローンで構成される可能性は低い。したがって、72μgのDNAでPCRを行う必要はありません。適切な商用キットを使用してDNAを分離します(「材料表」を参照)。100 ng/μL DNAを使用して、前に説明したプロトコル(セクション9.1.3)を使用して3-4 PCR反応を設定します。並べ替えられた細胞数が10万未満の場合、ゲノムDNA製剤の代わりに細胞ライセートを使用する。
- アリコートは、96ウェルPCRプレートで約10,000個の細胞/ウェルを提供します。
- プレート内の細胞をペレットにし、上清のほとんどを慎重に除去します。ペレットは見えません。
- 各井戸に25μLの水を加え、95°Cで10分間加熱します。
- 2 mg/mLの5 μLを各ウェルに1時間加え、56°Cでインキュベートします。その後、95°Cで10分間加熱し、プロテナーゼKを不活性化します。
- PCR反応ごとに10μLの細胞ライセート混合物を使用してください。ライセートは24時間以内に使用する必要があります。
10. インデックスのバーコード化とシーケンスのための PCR の第 2 ラウンド
- 第1ラウンドPCRから40 pg/μLに希釈します。
- サンプルごとに 1 つの PCR を設定します (表 7および8に記載されているガイドを使用してください)。高忠実度ポリメラーゼの使用は、sgRNA増幅中にポリメラーゼによってもたらされる誤差を最小限に抑えるために重要である。
- 2%(w/v)アガロースゲルで5μLのPCR産物を解決します。~330 bps で単一のクリアバンドが観察されるべきである。
- PCR製品に31.5 μL(全量0.7倍)の再懸濁ビーズを添加し、よく混合し、RTで5分間インキュベートすることで、PCR製品を精製します。
- チューブを磁気ラックに3分間置きます。ビーズはプレートの側面に取り込まれ、溶液は明確でなければなりません。上清を慎重に取り出して捨てます。
- 150 μlの80%の新しいエタノールをチューブに加えます。30sをインキュベートし、上清を慎重に取り出して捨てます。
- ステップ13.6を繰り返し、今度は180 μLで行います。その後、ビーズを5分間空気乾燥させます。
- マグネットからチューブを取り外します。ビーズから35 μLの無菌EBバッファーにDNAをエルテしたDNA標的。3分間インキュベートし、チューブを磁気ラックに戻して3分間入れます。
- 溶出したPCR産物を含む上清の約30μLをクリーンチューブに移します。
- 次世代シーケンシングプラットフォーム上でサンプルをシーケンスします。HumanV1 gRNA ライブラリの場合、表 4に示すカスタム プライマーを使用して、シーケンス 19 bp を行います。
11. 受容体と関連経路を同定するためのバイオインフォマティクス解析
- ソートされた母集団とソートされていない母集団から、MAGeCK のカウント関数を使用して参照ライブラリにシーケンスをマップします。この関数は、生のカウント ファイル (補足表 1) を生成します。
注: MAGeCK のインストールと MAGeCK 内のさまざまな機能の使用方法に関する詳細な手順については、 以前に公開された Wang ら20のプロトコルで説明されています。 - 画面で使用されている制御ライブラリの技術標準を確認します。
- 中央値は生のカウントを正規化し、R21または同等のソフトウェアのggplot2パッケージを使用して、プラスミド内のカウントの経験的累積密度関数プロットをプロットし、未ソートサンプルを制御する(図5A)。
- プラスミド集団からのカウントを「制御」として、並べ替えられていない制御サンプルからのカウントを「テスト」サンプルとして使用して、MAGeCK の-test関数を実行します。機能は遺伝子要約ファイル(補足表1)を生成することにある。
- 遺伝子サマリーファイルを開き、以前に分類された必須遺伝子と非必須遺伝子22に対するログ折りたたみ変更(neg|lfcカラム)の分布を描画します(図5B)。
- 著しく枯渇した遺伝子(neg|fdr < 0.05)を選択し、R(図5C)のエンリッチャー23パッケージまたは同等の経路濃縮パッケージを使用して経路濃縮解析を行う。
- デフォルト設定で MAGeCK の-test関数を実行します。未ソートのコントロール サンプルの生のカウントを "コントロール" として使用し、分析を実行するときに、並べ替えられたサンプルから "処理" としてカウントします。
- MAGeCK によって生成された遺伝子サマリー ファイルを開き、pos|rank列を昇順でランク付けします。ヒットの識別のカットオフとしてFDR (pos|fdr列) < 0.05 を使用します。受容体は、通常、高いランクで、しばしば最初の位置にある。
- R または同等のソフトウェアで正の選択(pos|score)のロバスト ランク付けアルゴリズム (RRA) スコアをプロットします (図 5D)。
- 遺伝子ヒットを選択し、濃縮経路を同定するために、濃縮物パッケージまたはR内の任意の同等の経路濃縮パッケージを使用して経路濃縮解析を行います。
結果
NCI-SNU-1およびHEK293細胞でそれぞれ行われるヒトTNFSF9およびP.ファルシパーラムRH5の結合パートナーの同定のための2つの代表的なゲノムスケールノックアウト画面からのシーケンシングデータがそれぞれ提供される(補足表1)。RH5の結合挙動は、ヘパラン硫酸とその既知の受容体BSG24(図3C)の両方によって影響を受けたのに対し、TNFRSF9は、その既知の受容体TNFSF9に特異的に結合し、可溶性ヘパリンとの導入時に結合を失わなかった。図3BBのタンパク質3はTNFRSF9を表す。
両方の細胞株について、3日後の制御変異体ライブラリーにおけるgRNAの分布(9、14、および16日後の転帰)も提供される(補足表1)。gRNA分布により、実験の過程を通してライブラリの複雑さが維持されていたことが明らかになった(図5A)。TNFSF9用リガンドの同定のための遺伝子スクリーンを14日目のポストトランスダクションで行ったのに対し、RH5については9日目のポストトランスダクションを行った。画面の技術的品質は、必須遺伝子22の基準集合に対する分布と比較して、非必須遺伝子の基準集合を標的とするgRNAの観察された折り畳み変化の分布を調べることによって評価された(図5B)。さらに、経路レベルの濃縮は、対照サンプルを元のプラスミドライブラリと比較する際に、期待される必須経路が同定され、「ドロップアウト」集団において有意に富化されたことも明らかにした。14 日目の NCI-SNU-1 サンプルの例を図 5Cに示します。
コントロールとソートされた集団におけるgRNAの分布は、MAGeCKの-test関数を用いて(MAGeCKからの遺伝子要約出力については補足表1を参照)、表現型スクリーンから受容体を同定するために使用した。遺伝子レベル解析においてMAGeCKによって報告された修飾されたRRAスコアは、p値でランク付けされた遺伝子に対してプロットされる。MAGeCK の RRA スコアは、gRNA が予想よりも一貫して高くランク付けされるメジャーを提供します。TNFRSF9 の画面では、トップ ヒットは TNFRSF9 の既知の結合パートナーである TNFSF9 (図 5D)でした。また、TP53経路に関連する多数の遺伝子も同定された。RH5の場合、既知の受容体(BSG)および硫酸化GAGs(SLC35B2)の産生に必要な遺伝子にSLC35B2加えて、追加の遺伝子(SLC16A1)も同定された(図5E)。SLC16A1は、細胞25の表面にBSGを密売するために必要なシャペロンである。これらの結果は、機能的な形態で細胞の表面に発現させるために必要な直接相互作用受容体および細胞成分を同定するスクリーンの能力を示す。

図1:細胞表面受容体を同定するための遺伝子スクリーニングアプローチの概要このアッセイは、3つの主要なステップで構成されています:まず、細胞表面受容体の異性ドメインを表す組換えタンパク質は、HEK293細胞のような構造的に重要な翻訳後修飾を加えることができる細胞株で発現される。単量体タンパク質のエクトドメインは、結合性を高めるためにストレプトアビジン-PEに結合することによってオリゴマー化される。第二に、これらの熱心なプローブは、陰性対照タンパク質(黒)と比較してPE蛍光(緑色)の顕著なシフトによって示される細胞株上の明るい染色が細胞表面結合パートナーの存在を示す細胞結合アッセイに使用される。第3に、受容体陽性Cas9発現細胞株が選択され、大多数のタンパク質コード遺伝子を標的としたgRNAを用いたゲノムスケールスクリーニングが行われる。変異体ライブラリーを生成する際には、ポアソン分布確率に基づく30%のトランスダクション効率を使用するのが一般的であり、結果的な表現型が特定のノックアウトに起因するように各細胞が単一のgRNAを受け取ることを保証する。トランスデューセされた細胞によって発現されるBFPマーカーは、FACSを用いてgRNAを含む細胞を選択するために使用される。フェノールチスクリーンは、転写後9〜16日の間に行われる。画面の日に、総変異細胞母集団は2つに分けられる。一方は対照集団として保持され、残りの半分は組換えタンパク質結合のために選択される。組換えタンパク質を結合できなくなった変異体ライブラリーの細胞はFACSを使用してソートされ、並べ替え対対照集団におけるgRNAの濃縮は、標識された熱心なプローブの細胞表面結合に必要な遺伝子を同定するために使用される。相当な時間を必要とするプロトコルのステップが示されます。この図はシャルマら19から変更されました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:ELISAベースの方法を用いて、ビオチン化タンパク質とストレプトアビジン-PEの比率を確立する。ビオチン化単量体タンパク質から熱心なプローブを生成するために用いられるストレプトアビジン-PE結合戦略の一例。ビオチン化モノマーの希釈系列をストレプトアビジンの固定濃度に対してインキュベートした。過剰なビオチン化モノマーを検出できない最小希釈は、ELISAによって決定した。ELISAは、10 ngのストレプトアビジン-PEを用いてタンパク質希釈液の範囲を前もって行った。ストレプトアビジン-PEの存在下で、シグナルが同定されなかった最小希釈(黒丸で囲まれた)と飽和に必要なタンパク質の量を計算し、4μg/mLストレプトアビジン-PEを有する10倍のストック溶液を生成した。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:細胞株へのタンパク質の代表的な結合。(A)細胞株に結合するタンパク質は、対照試料と比較して細胞関連蛍光が明らかに増加した。組換えタンパク質の熱処理(80°C、10分間10分間)は、全ての結合を陰性対照に戻し、正しく折り畳まれたタンパク質に結合挙動が依存していることを実証した。(B) 細胞表面に対するタンパク質結合行動の異なるクラス。GAGsへの依存。左から右へ、タンパク質は3つのタイプに分類することができます:タンパク質タイプ1はHSに吸い込むだけです。これらのタンパク質は、0.2 mg/mL以上のヘパリン濃度で前入りした後に結合を失います。タンパク質タイプ2は、特定の受容体に加えてHSに結合する。これらのタンパク質は、事前ブロッキング実験で部分的結合を失う。タンパク質タイプ3はHSに結合しない。これらのタンパク質は、親のラインと比較して結合を失いません.(C)添加された方法でHSおよび特異的受容体に結合するタンパク質(すなわち、RH5)の例。受容体(すなわち、BSG)またはHS合成に必要な酵素(例えば、SLC35B2、EXTL3)のいずれかを標的とすることは、コントロールに対する細胞へのRH5の結合を部分的に減少させるだけである。トランスデューセドポリクローナルラインは、結合挙動を確立するためにこのような実験に使用することができる。この図はシャルマら19から変更されました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:Cas9活性の高いクローン細胞株を選択する。NCI-SNU-1細胞株のポリクローナルおよびクローン化された細胞株のゲノム編集効率を、GFP-BFPレポーターシステムを用いて評価した。回路図が描かれています。フローサイトメトリーは、BFPとGFPの両方の発現をトランスダクション後にテストするために使用され、感染していない制御と比較した。GFP 式は Cas9 アクティビティのプロキシとして使用され、BFP 式はトランスデューシングされたセルをマークしました。感染していない細胞と空の感染細胞のプロファイルは、すべてのクローンに似ていました。代表的なプロファイルは、左側のパネルに示されています。NCI-SNU-1細胞株の5つのクローンすべてが、ポリクローナルライン(右パネル)に比べてGFPの損失が高く、クローン4は難治性の最も低い集団で最も高い効率を示した。この図はシャルマら19から変更されました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図5:細胞表面結合パートナーの同定のための遺伝子スクリーンからの代表的な結果。(A)9日目、14日目、16日目のプラスミドライブラリー中のgRNAの存在量をHEK-293-EおよびNCI-SNU-1細胞の変異ライブラリーと比較する累積分布関数プロット。任意の数値について、累積密度関数は、そのしきい値を下回ったデータポイントのパーセントを報告します。元のプラスミド集団と比較した変異細胞集団の小さなシフトは、プラスミドライブラリーと比較してgRNAのサブセットにおける枯渇を表す。(B) HEK293およびNCI-SNU-1細胞株において、これまで必須(赤)または非必須(黒)として分類された遺伝子におけるログフォールド変化の分布。~0に中心となる非必須遺伝子の折り目変化の分布は、必須遺伝子の場合は陰性の折り目変化に向かって左にシフトした。(C) NCI-SNU-1変異対照集団で枯渇した遺伝子の中で有意に富化した経路伝達後14日。期待される既知の細胞必須経路が同定された。(D) TNFRSF9プローブを結合する能力を失った分類された細胞内で富化された遺伝子のロバストランクアルゴリズム(RRA)スコア。遺伝子はRRAスコアに従ってランク付けされた。既知の相互作用パートナーTNFSF9およびTP53経路に関連する遺伝子(赤色で標識)が画面で同定された。(E) HEK293細胞に結合するRH5に必要なgRNA濃縮解析から同定された遺伝子に対するランク順のRRAスコア(左パネル)。SLC35B2およびSLC16A1は、5% の偽検出率 (FDR) しきい値内で識別されました。HS生合成経路における2つの追加遺伝子(すなわち、EXTL3およびNDST1)は、FDR内で25%の同定された。関連する遺伝子を対応するステップにマッピングした一般的なGAG生合成経路を示す回路図(パネル2)。コンドロイチン硫酸生物生成に必要な遺伝子(すなわち、CSGALNACT1/2)は画面で同定されなかった。この図はシャルマら19から変更されました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
| プラスミド名 | プラスミッド# | 使用 |
| タンパク質発現構築物:CD200RCD4d3+4-バイオリンカー-彼 | 追加遺伝子: 36153 | CD4d3+4、ビオチン、6-hisタグを用いた組換えタンパク質の製造 |
| pMD2.G | アドジーン: 12259 | プラスミドを発現するVSV-Gエンベロープ;レンチウイルスの生産 |
| psPAX2 | 追加遺伝子: 12260 | レンチウイルス包装プラスミド、レンチウイルスの製造 |
| キャス9-コンストラクト: pKLV2-EF1a-Cas9Bsd-W | アドジーン: 68343 | Cas9ラインを構成的に表現する生産 |
| gRNA発現構築物: pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W | アドジーン: 67974 | 改良された足場およびプロ/BFPマーカーを用いたCRISPR gRNA発現ベクター |
| ヒト改善ゲノムワイドノックアウトCRISPRライブラリ | アドジーン: 67989 | レンチウイルスでの使用のために設計された18,010のヒト遺伝子に対するgRNAライブラリー。 |
| GFP-BFP コンストラクト: pKLV2-U6gRNA5(gGFP)-PGKBFP2AGFP-W | アドジーン: 67980 | BFP と GFP を使用した Cas9 アクティビティ レポーター。 |
| 空の構造: pKLV2-U6gRNA5(空)-PGKBFP2AGFP-W | アドジーン: 67979 | BFP および GFP を使用した Cas9 アクティビティ レポーター (コントロール)。 |
表1:このアプローチで使用されるプラスミド。
| バッファ名 | コンポーネント |
| HBS (10X) | ミリク水の1.5 M NaClおよび200 mM HEPES、pH 7.4に合わせる |
| PBS (10X) | 80 g NaCl, 2 g KCl, 14.4 g Na2HPO4および 2.4 g KH2PO4ミリQ水で, pH 7.4 に調整 |
| リン酸ナトリウムバッファー(80mM在庫) | 7.1 g Na2HPO4.2H2O, 5.55 g NaH2PO4, pH 7.4 に調整 |
| His精製結合バッファー | 20 mM リン酸ナトリウムバッファー、0.5 M NaCl および 20 mM イミダゾール、pH 7.4 に調整 |
| ヒス精製溶出バッファー | 20 mM リン酸ナトリウムバッファー、0.5M NaCl および 400 mM イミダゾール、pH 7.4 に調整 |
| ジエタノールアミンバッファー | ミリQ水の10%ジエタノールアミンと0.5 mM MgCl2、pH 9.2に調整します: |
| D10 | DMEM、1%ペニシリンストレプトマイシン(100単位/mL)および10%の熱不活化FBS |
表2:この研究に必要なバッファ。
| コンポーネント | 10cm皿 | 6ウェルプレート |
| 293FT細胞 | コンフルエント 70~80% | コンフルエント 70~80% |
| トランスフェクション対応メディア(オプティ-MEM)(ステップ5.1.2) | 3 mL | 500 μL |
| トランスフェクション対応メディア(オプティ-MEM)(ステップ5.1.4) | 5 mL | 2 mL |
| レンチウイルス伝達ベクター | 3 μg | 0.5 μg |
| psPax2 (表 1 を参照) | 7.4 μg | 1.2 μg |
| pMD2.G (表 1 を参照) | 1.6 μg | 0.25 μg |
| プラス試薬 | 12 μL | 2 μL |
| リポフェクタミン LTX | 36 μL | 6 μL |
| D10 (ステップ 7.1.7) | 5 mL | 1.5 mL |
| D10 (ステップ 7.1.8 および 7.1.10) | 8 mL | 2 mL |
表3:レンチウイルス包装ミックスの試薬の量と量。
表4:gRNA及びNGSを増幅するためのプライマー配列。このファイルを表示するには、ここをクリックしてください (右クリックしてダウンロードしてください)。
| 試薬 | 反応あたりの体積 | マスターミックス (x38) |
| Q5 ホットスタート ハイフィデリティ 2x | 25 μL | 950 μL |
| プライマー(L1/U1)ミックス(各10μM) | 1 μL | 38 μL |
| ゲノムDNA (1 mg/mL) | 2 μL | 72 μL |
| H2O | 22 μL | 1100 μL |
| 合計 | 50 μL | 1900 μL |
表5:高複雑性サンプルからのgRNAの増幅のためのPCR。
| サイクル番号 | 変性 | 焼鈍 | 拡張子 |
| 1 | 98°C、30年代 | ||
| 2-24 | 98°C、10s | 61°C、15s | 72°C、20年代 |
| 25 | 72°C、2分 |
表6:第1PCRのPCR条件
| 試薬 | 反応あたりの体積 |
| KAPA ハイフィ ホットスタート レディミックス | 25 μL |
| プライマー(PE1.0/インデックスプライマー)ミックス(各5μM) | 2μL |
| 初の PCR 製品 (40 pg/μL) | 5 μL |
| H2O | 18 μL |
| 合計 | 50 μL |
表7:遺伝子スクリーンからのsgRNAのインデックスタグ付けに関するPCR。
| サイクル番号 | 変性 | 焼鈍 | 拡張子 |
| 1 | 98°C、30年代 | ||
| 2-15 | 98°C、10s | 66°C、15s | 72°C、20年代 |
| 16 | 72°C、5分 |
表 8: 第 2 PCR の PCR 条件

補助図 S1: 拘束できない母集団を並べ替えるゲートを描画するためのガイド。(A)スクリーニングのための理想的なタンパク質候補は、対照集団と比較して結合集団の明確なシフトを有し、HS生合成のための機械を欠く細胞に結合を保持すべきである。ヘパリンブロッキング実験は、SLC35B2標的細胞株での試験の代わりに使用することができる。(B)タンパク質外界から表面染色を欠いているがレンチウイルス導入からBFP蛍光を発現する細胞を採取した。表示される細胞は、GABBR222の受容体の同定のための画面からである。この図はシャルマら19から変更されました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

補足図S2:細胞表面糖タンパク質転写法は、1,000以上の癌細胞株からのRNA-seqデータを用いたPCAプロットに基づく。細胞モデルパスポート27の細胞株は、約1,500個の細胞表面糖タンパク質のFPKM値に従ってK-平均クラスタリングを用いてクラスター化した。各クラスターの代表的なセルラインにはラベルが付けられます。クラスター5は、造血起源の細胞株から完全に構成された(補足表2も参照)。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

補足図S3:プロジェクトスコアからのKEGG注釈タンパク質輸出およびN結合型グリコシル化遺伝子の必須性スコア。約330細胞株(カラム、標識なし)の調節されたベイズ必須性スコアは、タンパク質輸出およびN結合型グリコシル化経路(X軸)の遺伝子についてプロットされる。0より高いスコアは、元のプラスミドライブラリと比較して、突然変異体集団における有意な枯渇を表す。遺伝子は、細胞株の異なるレベルの本質的性を表す3つの異なるクラスターに分けることができます。このクラスタリングを使用して、ソートの日を決定できます。遅い時点(16日目)に画面が行われる場合、細胞に必須であることが知られている遺伝子(クラスター1および3)が同定されない可能性がある。この図の大きなバージョンを表示するには、ここをクリックしてください。
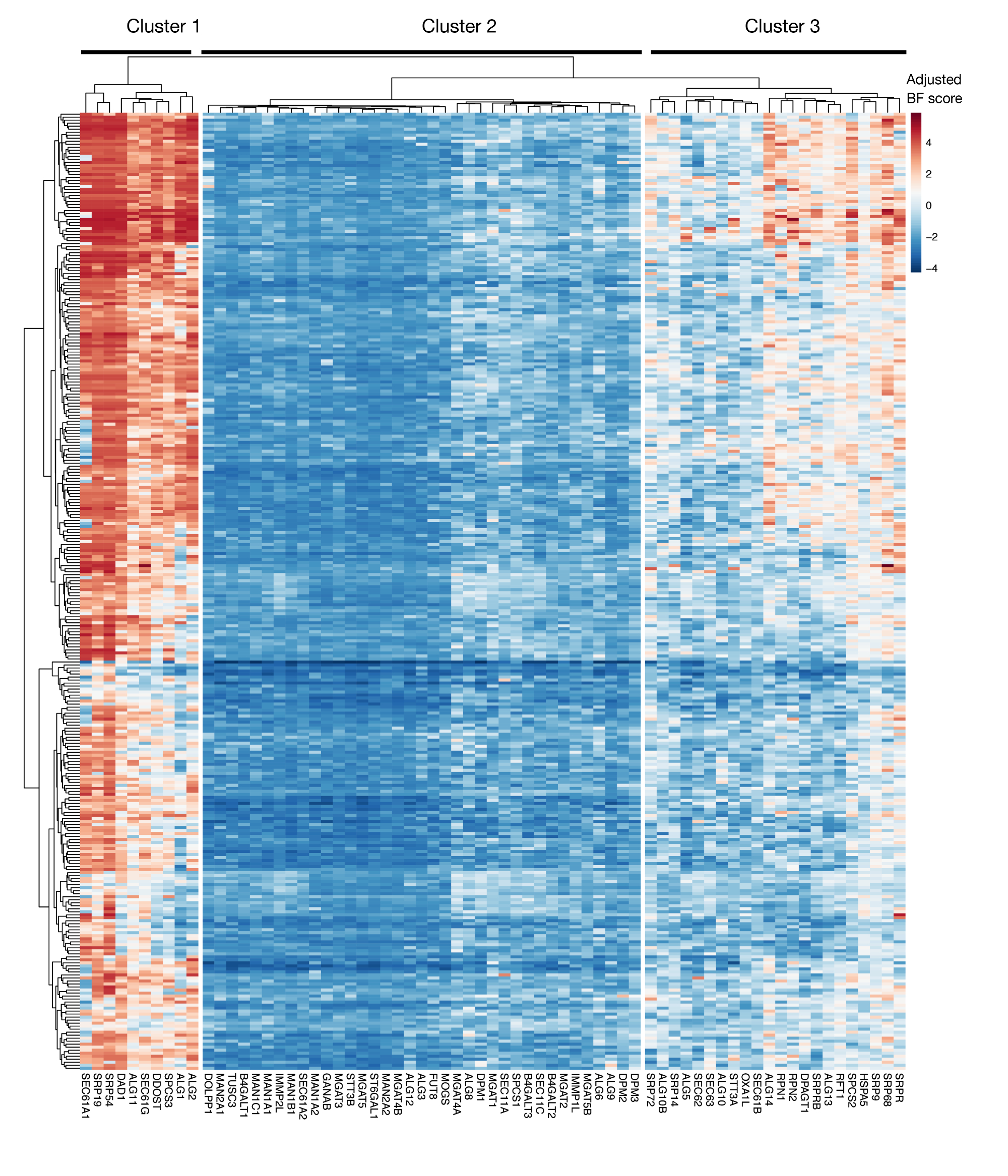{kind=link}
補足表1:代表的な遺伝的画面に関連するgene_summaryファイルを生成したMAGeCKソフトウェアとMAGeCKソフトウェアの生のカウントファイル。このファイルを表示するには、ここをクリックしてください (右クリックしてダウンロードしてください)。
補足表2:細胞表面受容体の発現に応じた細胞株のクラスタリングこのファイルを表示するには、ここをクリックしてください (右クリックしてダウンロードしてください)。
ディスカッション
細胞認識に関与する細胞成分をコードする遺伝子を同定するCRISPRベースのスクリーニング戦略について説明する。CRISPR活性化を用いる同様のアプローチは、大きなタンパク質ライブラリー26を生成する必要なしに組換えタンパク質の直接相互作用受容体を同定する遺伝的代替手段も提供する。しかし、このアプローチの1つの大きな利点は、細胞にネイティブに表示される表面分子によって媒介される相互作用に適用可能であり、受容体の過剰発現に依存しないことであり、受容体の結合的な有数に影響を及ぼす可能性がある。したがって、他の方法とは異なり、この技術は、受容体の生化学的性質または細胞生物学に関する仮定を行うものではなく、非常に大きなタンパク質、または膜を複数回横断したり、他のタンパク質と複合体を形成したりするタンパク質、およびグリカン、糖脂質、およびリン脂質などのタンパク質以外の分子を使用して通常研究が困難なタンパク質によって媒介される相互作用を研究する機会を提供する。この手法のゲノムスケールの性質を考えると、このアプローチは、受容体を同定するだけでなく、結合事象に必要な追加の細胞成分を同定する利点もあり、それによって受容体の細胞生物学に関する洞察を提供する。
この方法を使用して孤立タンパク質の受容体を同定する際の主な制限の1つは、タンパク質に結合する細胞株を最初に同定するための最初の要件である。これは必ずしも容易ではなく、遺伝的スクリーンにも許容される結合表現型を表示する細胞株を同定することは、このアッセイを展開するための時間制限ステップとなり得る。一部の細胞株は、他のものよりも多くのタンパク質に結合する傾向があります。これは、HSに結合するタンパク質に特に関連します, これらのタンパク質は、HS側鎖を表示する任意の細胞株に結合する傾向があるため, ネイティブ結合コンテキストに関係なく.さらに、細胞株中のシンデカン(すなわち、HSを含有するプロテオグリカン)のアップレギュレーションがHS結合タンパク質26の結合の増加につながることを観察した。これは、スクリーニング用の細胞ラインを選択する際に考慮すべき要因である可能性があります。しかしながら、HSの添加結合が特異的受容体への結合を妨げないことも重要である。これは、結合が観察された場合、このアッセイにおいてHSによって媒介される結合が共依存性19ではなく添加剤であるため、HSのみによって媒介される可能性があることを意味する。このようなシナリオでは、記載されたヘパリン遮断アプローチは、完全な遺伝的画面を実行する必要なしにそのような行動を識別することができる。
細胞株を選択する有用なリソースは、ゲノム、トランスクリプトミクス、および約1,000の癌細胞株27の培養条件情報を含む細胞モデルパスポートである。生物学的文脈に応じて、細胞は発現プロファイルに基づいて選択することができる。細胞株の選択を支援するために、我々は、約1,500個のプリアナンツされたヒト細胞表面糖タンパク質28の発現に従って、細胞モデルパスポート内に~1,000個の細胞株を集集した(補足図2;成長条件と共に各細胞株のクラスター情報が補足表2に提供される)。未知の機能を有するタンパク質の結合をテストする場合、各クラスターから代表的な細胞株のパネルを選択して、幅広い受容体をカバーする可能性を高めると便利です。選択を与えられて、培養が容易で、トランスデュースが容易な細胞株を選ぶのが推奨される。これらの細胞株はゲノムスケールスクリーニングに用いるので、後のステップでCRISPRベースの遺伝子スクリーニングのためのsgRNAの送達に最も一般的に利用可能な方法であるため、大量に容易に増殖することができ、レンチウイルス伝達に寛容であることが好ましい。
一般的に、表現型の選択は単一のソートで行われます。しかし、これは、コントロールと比較して染色された細胞集団の明るさによって決定される。反復的な選択のラウンドは、目的の表現型の信号対雑音比が低い場合、または画面の目的が強い表現型を有する変異体を同定することである場合に採用される可能性がある。FACS ベースの画面に反復選択アプローチを使用する場合、選別プロセスは、主にソーターの完全な力のために、細胞死を引き起こす可能性があることを考慮することが重要です。したがって、収集されたすべてのセルが次の並べ替えのラウンドで表されるわけではありません。
ライブラリの複雑さは、特にネガティブな選択画面の場合、結果を開始ライブラリに存在していたものと比較することによってのみ決定できるため、遺伝的画面を成功させる上で非常に重要な要素です。否定的な選択画面では、500-1,000 x の複雑さのライブラリを維持するのが一般的です。しかし、正の選択画面は、特定の表現型に対して少数の変異体しか選択されないため、ライブラリサイズに対してより堅牢である。したがって、ここで説明する正の選択画面では、画面の品質を損なうことなくライブラリサイズを50〜100倍の複雑さに縮小することができる。さらに、これらの画面では、特定のセルラインの特定の日にコントロールライブラリを、その特定のセルラインに対してその日にソートされたすべてのサンプルの「一般的なコントロール」として使用することもできます。これにより、作成およびシーケンス処理が必要なコントロール ライブラリの数が減ります。
このアプローチを使用するためのもう一つの重要な考慮事項は、インビトロ細胞増殖に不可欠な遺伝子を同定する際の機能喪失スクリーンの限界です。この点では、変異細胞が培養中に保持される時間が長いほど、必須遺伝子に変異を有する細胞が生存不能になり、変異ライブラリーにもはや表現されない可能性が高くなる。300以上の細胞株におけるプロジェクトスコアイニシアチブの一部として行われた最近の遺伝的スクリーンは、KEGGアクローゼンタンパク質分泌およびN-グリコシル化経路における複数の遺伝子が、多くの場合、多くの細胞株に不可欠であると同定されることを示している(補足図3)29。細胞認識プロセスの文脈で増殖と生存に必要な遺伝子の効果を調査する場合、スクリーンを設計する際に考慮することができます。この場合、早期の時点(例えば、9日目のポストトランスダクション)で画面を実施することは、一般的に適切であろう。しかし、このアプローチを使用して、一般的な細胞経路ではなく、強いサイズ効果を持ついくつかのターゲットを同定する場合、後の時点(例えば、15-16日のポストトランスダクション)で画面を実行することが適切である可能性があります。
スクリーニングからの結果は非常に強い;過去に行われた8つの組換えタンパク質結合スクリーンにおいて、細胞表面受容体は、すべての場合19においてトップヒットした。このアプローチを使用して相互作用パートナーを同定する場合、受容体と表面上の提示に寄与する因子が高い統計的信頼度で識別されることを期待すべきである。画面が実行され、単一のgRNAノックアウトを使用してヒットが検証されると、AVEXIS4などの既存の生化学的方法と表面プラズモン共鳴を使用した精製タンパク質の直接焼成可能な結合を使用して、さらなるフォローアップを行うことができます。ここで説明するアプローチは、可溶型組換え結合プローブを生成することができるすべてのタンパク質に適用可能である。
要約すると、これは細胞表面タンパク質によって媒介される相互作用を同定するためのゲノムスケールCRISPRノックアウトアプローチである。この方法は、一般に、生物自身の細胞間(例えば、神経および免疫学的認識)、ならびに宿主細胞と病原体タンパク質の間を含む、幅広い異なる生物学的文脈における細胞表面認識に必要な細胞経路を同定するために適用可能である。この方法は、受容体同定のために設計された生化学的アプローチに代わる遺伝的代替手段を提供し、受容体の生化学的性質または細胞生物学に関する事前の仮定を必要としないため、完全に予期せぬ発見を行う大きな可能性を秘めている。
開示事項
著者らは開示するものは何もない。
謝辞
この作品は、GJWに授与されたウェルカムトラスト助成金番号206194によってサポートされました。私たちは、サイトメトリーコア施設に感謝します:ビーリンン、ジェニファーグラハム、サムトンプソン、クリストファーホールはFACSの助けを借りています。
資料
| Name | Company | Catalog Number | Comments |
| Anti-mouse alkaline phosphatase | Sigma | A4656 | |
| Blasticidin | Chem-Cruz | SC-204655 | |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | 13362 | |
| BSA | Sigma | A9647-100G | |
| Diethanolamine | Sigma | 398179 | |
| DMEM | Gibco | 31966-021 | |
| Dneasy Blood and Tissue kit | Qiagen | 69504 | |
| DynaMag-96 Side Magnet | Invitrogen | 12331D | |
| HEK293T packaging cells | ATCC | CRL-3216 | |
| Heparin | Sigma | H4784-1G | |
| KAPA HiFi HotStart ReadyMix | Kapa | KK2602 | |
| Lipofectamine LTX with PLUS reagent | Invitrogen | 15338100 | |
| MoFlo XDP cell sorter | BD | ||
| Ni2+-NTA agarose beads | Jena Bioscience | AC-501-25 | |
| OPTI-MEM | Life Technologies | 31985-070 | |
| OX-68 antibody | AbD Serotec | MCA1022R | |
| p-nitrophenyl phosphate | Sigma | 1040-506 | |
| PD-10 desalting columns | GE healthcare | 17085101 | |
| Polybrene | Millipore | TR-1003-G | |
| Polypropylene tubes with 5 mL bed volume | Qiagen | 34964 | |
| Proteinase K, recombinant, PCR Grade | Roche | 3115879001 | |
| Puromycin | Gibco | A11138-03 | |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | M0494L | |
| QIAquick PCR purification kit | Qiagen | 28104 | |
| SCFA filter | Nalgene | 190-2545 | |
| Sony Cell sorter | Sony | ||
| SPRI beads (Agencourt AMPure XP beads) | Beckman | A63881 | |
| Streptavidin-coated microtitre plates | Nalgene | 734-1284 | |
| Streptavidin-PE | Biolegend | 405204 |
参考文献
- Wright, G. J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems. 5 (12), 1405-1412 (2009).
- van der Merwe, P. A., Barclay, A. N. Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends in Biochemical Sciences. 19 (9), 354-358 (1994).
- Wood, L., Wright, G. J. Approaches to identify extracellular receptor-ligand interactions. Current Opinion in Structural Biology. 56, 28-36 (2019).
- Bushell, K. M., Söllner, C., Schuster-Boeckler, B., Bateman, A., Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Research. 18 (4), 622-630 (2008).
- Visser, J. J., et al. An extracellular biochemical screen reveals that FLRTs and Unc5s mediate neuronal subtype recognition in the retina. eLife. 4, e08149(2015).
- Özkan, E., et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell. 154 (1), 228-239 (2013).
- Martinez-Martin, N., et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell. 174 (5), 1158-1171 (2018).
- Bianchi, E., Doe, B., Goulding, D., Wright, G. J. Juno is the egg Izumo receptor and is essential for mammalian fertilization. Nature. 508 (7497), 483-487 (2014).
- Mullican, S. E., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature Medicine. 23 (10), 1150-1157 (2017).
- Turner, L., et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 498 (7455), 502-505 (2013).
- Frei, A. P., et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nature Biotechnology. 30 (10), 997-1001 (2012).
- Sobotzki, N., et al. HATRIC-based identification of receptors for orphan ligands. Nature Communications. 9 (1), 1519(2018).
- Sharma, S., Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 19 (4), (2018).
- Gebre, M., Nomburg, J. L., Gewurz, B. E. CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses. 10 (2), (2018).
- Zotova, A., Zotov, I., Filatov, A., Mazurov, D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. Journal of Immunological Methods. 439, 8-14 (2016).
- Puschnik, A. S., Majzoub, K., Ooi, Y. S., Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nature Reviews. Microbiology. 15 (6), 351-364 (2017).
- Kerr, J. S., Wright, G. J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. Journal of Visualized Experiments. (61), e3881(2012).
- Tzelepis, K., et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Reports. 17 (4), 1193-1205 (2016).
- Sharma, S., Bartholdson, S. J., Couch, A. C. M., Yusa, K., Wright, G. J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Research. 28 (9), 1372-1382 (2018).
- Wang, B., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nature Protocols. 14 (3), 756-780 (2019).
- R Core team. A language and environment for statistical computing. , http://www.R-project.org (2013).
- Hart, T., et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3. 7 (8), 2719-2727 (2017).
- Kuleshov, M. V., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44 (W1), W90-W97 (2016).
- Crosnier, C., et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 480 (7378), 534-537 (2011).
- Kirk, P., et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal. 19 (15), 3896-3904 (2000).
- Chong, Z. S., Ohnishi, S., Yusa, K., Wright, G. J. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biology. 19 (1), 205(2018).
- van der Meer, D., et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Research. 47 (D1), D923-D929 (2019).
- Bausch-Fluck, D., et al. A mass spectrometric-derived cell surface protein atlas. PloS One. 10 (3), e0121314(2015).
- Behan, F. M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 568 (7753), 511-516 (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved