
Method Article
Identification des récepteurs de surface cellulaire à l’aide d’écrans génétiques CRISPR/Cas9 à l’échelle du génome
Dans cet article
Résumé
Ce manuscrit décrit une approche de dépistage à l’échelle du génome basée sur les cellules pour identifier les interactions extracellulaires de récepteur-ligand.
Résumé
La communication intercellulaire médiée par des interactions directes entre les récepteurs de surface des cellules intégrées par la membrane est cruciale pour le développement normal et le fonctionnement des organismes multicellulaires. La détection de ces interactions reste toutefois techniquement difficile. Ce manuscrit décrit une approche systématique de dépistage génétique CRISPR/Cas9 à l’échelle du génome qui révèle les voies cellulaires requises pour des événements spécifiques de reconnaissance de surface cellulaire. Cet essai utilise des protéines recombinantes produites dans un système d’expression protéique des mammifères comme sondes de liaison avides pour identifier les partenaires d’interaction dans un écran génétique à base de cellules. Cette méthode peut être utilisée pour identifier les gènes nécessaires aux interactions de surface cellulaire détectées par des sondes liants recombinantes correspondant aux ectodomains des récepteurs membranaires. Fait important, étant donné la nature à l’échelle du génome de cette approche, il a également l’avantage non seulement d’identifier le récepteur direct, mais aussi les composants cellulaires qui sont nécessaires pour la présentation du récepteur à la surface cellulaire, fournissant ainsi des informations précieuses sur la biologie du récepteur.
Introduction
Les interactions extracellulaires par les protéines des récepteurs de surface cellulaire dirigent des processus biologiques importants tels que l’organisation des tissus, la reconnaissance hôte-pathogène et la régulation immunitaire. L’étude de ces interactions est d’intérêt pour l’ensemble de la communauté biomédicale, parce que les récepteurs membranaires sont des cibles exploitables de thérapeutiques systématiquement livrés tels que les anticorps monoclonaux. Malgré leur importance, l’étude de ces interactions reste techniquement difficile. C’est principalement parce que les récepteurs membranés sont amphipathiques, ce qui les rend difficiles à isoler des membranes biologiques pour la manipulation biochimique, et leurs interactions sont caractérisées par les affinités d’interaction faibles (KDs dans la gamme M-mM)1. Par conséquent, de nombreuses méthodes couramment utilisées ne sont pas adaptées pour détecter cette classe d’interactions protéiques1,2.
Une gamme de méthodes a été développée pour étudier spécifiquement les interactions extracellulaires de récepteur-ligand qui prennent leurs propriétés biochimiques uniques en considération3. Un certain nombre de ces approches consistent à exprimer l’ectodomain entier d’un récepteur comme protéine recombinante soluble dans les systèmes à base de mammifères ou d’insectes à base de cellules pour s’assurer que ces protéines contiennent des modifications posttranslationnelles qui sont structurellement importantes, telles que les glycanes et les liens de disulfide. Pour surmonter la liaison de faible affinité, les ectodomains sont souvent oligomés pour augmenter leur avidité liante. Les ectodomains de protéine avide ont été utilisés avec succès comme sondes contraignantes pour identifier les partenaires d’interaction dans les écrans d’interaction protéine-protéine recombinants directs4,5,6,7. Bien que largement réussies, les méthodes recombinantes à base de protéines exigent que l’ectodomain d’un récepteur membranaire soit produit comme protéine soluble. Par conséquent, il ne s’applique généralement qu’aux protéines qui contiennent une région extracellulaire contigue (p. ex., le type I, le type II ou l’IPG) et ne convient généralement pas aux complexes de récepteurs et aux protéines membranaires qui couvrent la membrane plusieurs fois.
Les techniques de clonage d’expression dans lesquelles une bibliothèque de DNA complémentaires (ACS) est transposée en cellules et testées pour un phénotype de gain-of-binding ont également été employées pour identifier les interactions extracellulaires protéine-protéine8. La disponibilité de grandes collections de plasmides d’expression cDNA clonés et séquencés ces dernières années a facilité les méthodes dans lesquelles les lignées cellulaires surexprimant les CDNA codant les récepteurs de surface cellulaire sont examinés pour la liaison des protéines recombinantes pour identifier les interactions9,10. Les approches basées sur la cDNA, contrairement aux méthodes recombinantes à base de protéines, offrent la possibilité d’identifier les interactions dans le contexte de la membrane plasmatique. Cependant, le succès de l’utilisation des constructions d’expression cDNA dépend de la capacité des cellules à surexprimer la protéine sous la forme correctement pliée, mais cela nécessite souvent des facteurs d’accessoires cellulaires tels que les transporteurs, les chaperons, et l’assemblage oligomeric correct. Transfecter une seule CDNA pourrait donc ne pas suffire à obtenir l’expression de la surface cellulaire.
Les techniques de criblage à l’aide de constructions d’ADN ou de sondes de protéines recombinantes exigent de grandes collections de bibliothèques de protéines cDNA ou recombinantes. Des méthodes basées sur la spectrométrie de masse spécialement conçues ont été utilisées récemment pour identifier les interactions extracellulaires qui ne nécessitent pas l’assemblage de grandes bibliothèques. Cependant, ces techniques nécessitent une manipulation chimique de la surface cellulaire, qui peut modifier la nature biochimique des molécules présentes à la surface des cellules et ne sont actuellement applicables que pour les interactions négociées par des protéines glycosylated11,12. La majorité des méthodes actuellement disponibles se concentrent également fortement sur les interactions entre les protéines tout en ignorant largement la contribution du microenvironnement membranaire, y compris des molécules telles que les glycanes, les lipides et le cholestérol.
Le développement récent d’un ciblage bialléléique très efficace à l’aide d’approches basées sur CRISPR a permis aux bibliothèques à l’échelle du génome de cellules dépourvues de gènes définis dans un seul bassin qui peut être examiné de manière systématique et impartiale pour identifier les composants cellulaires impliqués dans différents contextes, y compris la dissection des processus de signalisation cellulaire, l’identification des perturbations qui confèrent une résistance aux médicaments, toxines et agents pathogènes, et la détermination de la spécificité des anticorps13,14,15,16. Ici, nous décrivons un essai de dépistage par KO à l’échelle du génome à base de CRISPR qui offre une alternative aux approches biochimiques actuelles pour identifier les interactions extracellulaires de récepteur-ligand. Cette approche d’identification des interactions négociées par les récepteurs membranaires par des écrans génétiques est particulièrement adaptée aux chercheurs qui ont un intérêt ciblé sur les ligands individuels, car il évite la nécessité de compiler de grandes bibliothèques de cDNA ou de protéines recombinantes.
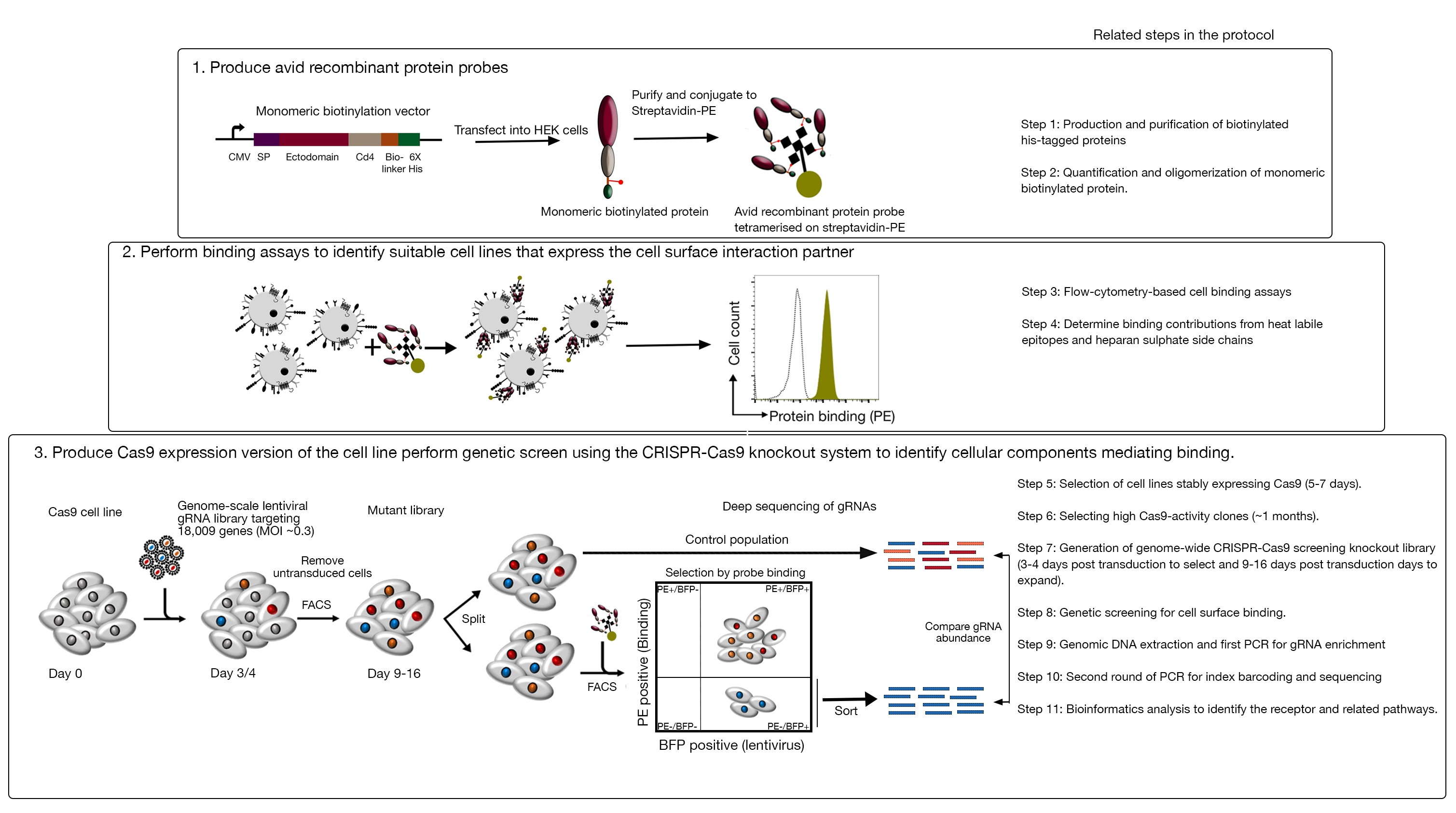
Cet essai se compose de trois étapes majeures : 1) Des sondes de liaison de protéine recombinantes très avides composées des régions extracellulaires d’un récepteur d’intérêt sont produites et utilisées dans les essais de liaison à base de cytométrie de débit à base de fluorescence ; 2) Les essais de liaison sont employés pour identifier une ligne cellulaire qui exprime le partenaire d’interaction de la sonde de protéine recombinante ; 3) Une version Cas9-exprimant de la lignée cellulaire qui interagit avec la protéine d’intérêt est produite et un écran à élimination directe à l’échelle du génome CRISPR/Cas9 est effectué(figure 1). Dans cet écran génétique, la liaison d’une protéine recombinante aux lignées cellulaires est utilisée comme phénotype mesurable dans lequel les cellules de la bibliothèque knock-out qui ont perdu la capacité de lier la sonde sont triées à l’aide de tri cellulaire activé à base de fluorescence (FACS) et les gènes qui ont causé la perte du phénotype de liaison identifié par le séquençage. En principe, les gènes codant le récepteur responsable de la liaison de la sonde avide et ceux requis pour son affichage de surface cellulaire sont identifiés.
La première étape de ce protocole implique la production de sondes de protéines recombinantes avides représentant l’ectodomain des récepteurs liés à la membrane. Ces récepteurs sont connus pour conserver fréquemment leurs fonctions de liaison extracellulaire lorsque leurs ectodomains sont exprimés comme une protéine soluble recombinante1. Pour une protéine d’intérêt, les protéines recombinantes solubles peuvent être produites dans n’importe quel système approprié d’expression des protéines eucaryotes dans n’importe quel format à condition qu’il puisse être oligoméized pour une avidité contraignante accrue, et il contient des étiquettes qui peuvent être utilisées dans les essais de liaison à base de cytométrie de flux à base de fluorescence (par exemple, FLAG-tag, biotin-tag). Des protocoles détaillés pour la production d’ectodomains solubles de récepteurs membranaires utilisant le système d’expression protéique HEK293, ainsi que différentes techniques de multimerisation et les constructions d’expression protéinées pour la production de protéines pentamériques et de protéines monomériques ont été précédemment décrites1,17. Le protocole ici décrira les étapes pour générer des sondes fluorescentes avides à partir de protéines biotinylated monomériques en les conjuguant à la streptavidin conjuguée à un fluorochrome (par exemple, la phycoerythrine, ou PE), qui peut être utilisé directement dans les essais de liaison à base de cellules et a l’avantage de ne pas avoir besoin d’un anticorps secondaire pour la détection. Les protocoles généraux pour l’exécution des écrans à l’échelle du génome ont déjà été décrits20,21, de ce fait, le protocole se concentre principalement sur les spécificités de l’exécution des écrans de liaison de protéines recombinantes à base de cytométrie d’écoulement à l’aide du système de dépistage à élimination directe CRISPR/Cas9 à l’aide de la bibliothèque human V1 (« Yusa »)18.
Protocole
1. Production et purification de protéines biotinées his-étiquetées
- Utilisez un système d’expression protéique à base de mammifères ou d’insectes pour produire des protéines biotinylateds solubles recombinantes étiquetées (voir les constructions de plasmides dans le tableau 1).
REMARQUE : Un protocole détaillé pour la production de biotine monomérique et de protéines his-étiquetées utilisant le système d’expression cellulaire HEK293 est décrit par Kerr et coll.17. Les ectodomains protéiques exprimés à l’aide du système d’expression HEK293 sont sécrétés dans le milieu de la culture. - Recueillir les protéines solubles en granuler les cellules par centrifugation à 3.000 x g pendant 20 min.
- Filtrer le supernatant à l’intermédiaire d’un filtre de 0,22 m et ajouter les perles d’agarose Ni2 'NTAau supernatant de protéine filtrée dans un rapport de 1:1,000 (c.-à-d. 50 l de boue agarose de 50 % en 50 ml de supernatant). Incuber pendant la nuit ou au moins 4-5 h à 4 oC sur une plate-forme tournante.
- Laver la colonne de polypropylène en ajoutant 5 ml de tampon de lavage de sa purification. Consultez le tableau 2 pour toutes les compositions tampons.
- Verser l’ensemble du mélange supernatant perle-protéine dans la colonne. Les perles s’accumuleront à la base.
- Laver les perles 2x avec 15 ml de tampon de lavage. Pour éviter la dilution des protéines, dessinez soigneusement le tampon de lavage résiduel de la colonne avec une seringue de 5 ml et jetez.
- Ajouter soigneusement 300-500 l de tampon d’elution de sa purification directement aux perles et incuber pendant au moins 1 h. Recueillir la protéine elfée en tirant à nouveau soigneusement le liquide à l’aide d’une seringue de 1 mL. Échangez le tampon d’elution au tampon désiré (p. ex., normalement PBS ou HBS) à l’aide de colonnes de désalting. Conserver toutes les protéines à 4 oC jusqu’à nouvel appel.
2. Quantification et oligomerisation de protéines biotinylated monoméaniques
REMARQUE : Pour augmenter l’avidité liante, oligomerize protéines monoméanes biotinylated sur streptavidin-PE tétramérique avant de les utiliser dans les essais contraignants. Atteindre des rapports de conjugaison optimaux de protéines monomériques et de streptavidine-PE tétramériques en testant une série de dilution de monomers biotinylés contre une concentration fixe de streptavidine et en établissant empiriquement la dilution minimale à laquelle aucun excès de monomers biotinylated ne peut être détecté.
- Faire au moins huit dilutions sérieuses d’échantillons de protéines biotinylated à l’aide d’un tampon de dilution approprié (PBS ou HBS avec 1% d’albumine de sérum bovin [BSA]) dans une plaque de puits de 96. Assurez-vous que le volume final de chaque dilution est d’au moins 200 ll.
- Faire une plaque en double des échantillons en enlevant 100 L de chaque puits et en transférant dans une nouvelle plaque de puits 96. Toujours inclure un contrôle. Dans ce cas, les contrôles sont des protéines tag-only (c.-à-d., biotinylé Son-t marqué Domaine Cd4 domaine 3-4 protéine). Ceci sera utilisé comme sonde de contrôle dans tous les essais de liaison.
- Diluer la streptavidine-PE à 0,1 g/mL dans le tampon de dilution.
- Sur une seule des plaques, ajouter 100 lil de la streptavidin-PE diluée. La plaque en double servira de contrôle. Ajouter 100 L de tampon de dilution dans la plaque de contrôle pour égaliser les volumes.
- Incuber pendant 20 min à température ambiante (RT). Pendant ce temps, bloquer les puits d’une plaque enduite de streptavidin avec le tampon de dilution pendant 15 min.
- Transférer le volume total de l’échantillon des deux plaques vers des puits individuels des plaques enduites de streptavidin et incuber pendant 1 h à RT.
- Laver la plaque 3x avec 200 L de tampon de lavage (c.-à-d., PBS ou HBS avec 0,1% Tween-20, 2% BSA). Ajouter 100 L de souris anti-rat cd4d3-4 IgG (OX68) et incuber pendant 1 h à RT.
- Laver la plaque 3x avec le tampon de lavage. Ajouter 100 l d’un anti-souris alkaline phosphatase conjugué à 0,2 g/mL pour 1 h à RT.
- Laver la plaque 3x avec tampon de lavage et 1x dans le tampon de dilution.
- Préparer le phosphate de p-nitrophenyl à 1 mg/mL dans le tampon de dénoolamine. Ajouter 100 ll dans chaque puits et incuber pendant 15 minutes.
- Prenez les lectures d’absorption à 405 nm. Utilisez la dilution minimale à laquelle il n’y a pas de signal sur la plaque comme facteur de dilution approprié pour créer des tétramers(figure 2).
- Faire une solution de colorant 10x pour tous les échantillons et contrôles en incuber 4 g/mL streptavidin-PE et la dilution protéinée biotinylated appropriée pendant 30 min chez RT. Stocker des protéines conjuguées dans un tube protégé par la lumière à 4 oC jusqu’à nouvel utilisation.
3. Flow cytométrie-basé sur les essais de liaison cellulaire
- Pour les cellules adhérentes, retirez les supports de culture et lavez 1x avec PBS sans cations divalentes. Ajoutez ensuite des solutions de décollement cellulaire (p. ex., EDTA). Laisser les cellules se détacher pendant 5-10 min. Appuyez doucement sur le flacon pour libérer les cellules.
REMARQUE : Évitez d’utiliser des produits à base de trypsine car ils peuvent fendre les protéines de surface des cellules. - Recueillir des cellules détachées dans un tube. Pour les cellules qui poussent en suspension (p. ex., cellules HEK293), collectez directement les cellules des flacons de culture dans un tube.
- Cellules de granule à 200 x g pendant 5 min. Retirez le supernatant et réutilisez la granule dans le tampon de lavage (c.-à-d., PBS/1% BSA).
- Comptez les cellules à l’aide d’un hémocytomètre et ajustez la concentration à 2,5 x 105x 106 cellules/mL. Aliquot 100 L de mélange cellulaire préparé sur une plaque à fond de 96 puits U ou V. Tourner la plaque pendant 5 min à 400 x g. Retirer le supernatant à l’égard d’une pipette multicanal.
- Ajoutez 100 L de sondes de protéines et de contrôles hautement avides étiquetés fluorescents normalisés dans les plaques préparées précédemment avec des cellules et incuber pendant 1 h à 4 oC. Après fixation pendant 1 h, faire tourner la plaque à 400 x g pendant 5 min.
- Retirez le supernatant et ajoutez 200 L de tampon de lavage (c.-à-d., PBS/1% BSA). Bien mélanger en faisant de la tuyauterie de haut en bas.
- Pelleter les cellules par centrifugation à 400 x g pendant 5 min. Répétez l’étape de lavage 1x. Après deux lavages, retirez complètement le supernatant et réutilisez la pastille de cellules dans 100 L de PBS.
- Analyser les cellules par cytométrie d’écoulement. Utilisez le laser jaune-vert (soit 561 nm) pour détecter la fluorescence PE.
- D’abord analyser les cellules qui ont été tachées avec la sonde de contrôle. Basé sur la distribution de la fluorescence PE, tracer une porte pour la population liante de telle sorte que pas plus de 1% de la cellule témoin tombe dans cette porte.
- Analyser l’échantillon et déterminer la fraction des cellules qui tombe dans la porte de liaison.
REMARQUE : Les lignées cellulaires qui affichent une population plus contraignante sont souhaitées pour les écrans génétiques, car elles ont un rapport signal-bruit plus élevé. Idéalement, plus de 80% des cellules devraient tomber dans cette porte.
4. Déterminer les contributions contraignantes des épitopes labiles thermiques et des fauteuils sidechains de sulfate de heparan
REMARQUE : L’activité de nombreuses protéines est labile de chaleur, de sorte que la perte d’activité de liaison après traitement thermique est encourageante. Il est conseillé de déterminer la contribution des glycosaminoglycans chargés négativement, principalement le sulfate de chaux (HS), dans la liaison de médiation des protéines recombinantes. C’est parce que la liaison par HS dans l’analyse de liaison cellulaire décrit ici peut être additif plutôt que codépendant sur d’autres récepteurs19. Cela signifie que la liaison observée pourrait être entièrement négociée par les chaînes latérales HS des protéoglycans de surface cellulaire et non par un récepteur spécifique. La liaison vers le HS à la surface cellulaire n’est pas nécessairement non spécifique, mais plutôt une propriété d’une protéine, ce qui est utile de savoir avant d’effectuer un écran génétique complet.
- Préparer des échantillons de protéines traités à la chaleur pour les utiliser dans les essais de liaison.
- Chauffer la protéine monomérique normalisée mais nonconjuguée à 80 oC pendant 10 min.
- Conjuguer la protéine traitée à la chaleur à la streptavidine-PE en supposant le même rapport de conjugaison que son homologue non traité tel que déterminé par ELISA (se référer à l’article 2).
- Préparer des échantillons de protéines bloqués par l’héparine.
- Préparer huit dilutions de 1:3 d’héparine soluble dans PBS avec une concentration de départ de 2 mg/mL et un volume final de 100 ll.
- Incuber 100 L de sondes de liaison préparées dans les dilutions de l’héparine pendant au moins 30 min.
- Utilisez des protéines traitées à la chaleur et le mélange complet de 200 l d’héparine/protéine dans les essais de liaison décrits à la section 3. Les résultats représentatifs sont présentés à la figure 3A,B.
5. Sélection des lignes cellulaires exprimant de façon stable Cas9
REMARQUE : Avant que la ligne cellulaire qui lie la sonde d’intérêt puisse être utilisée dans le criblage CRISPR, elle doit d’abord être conçue pour exprimer la nucléase Cas9 et un clone très actif sélectionné19.
- Utilisez le protocole de production général suivant de lentivirus pour produire le lentivirus à l’aide de la construction lentivirale pour l’expression Cas9 (se référer au tableau 1).
- Cellules de culture HEK293-FT dans les médias DMEM/10% FBS à 37 oC et 5% DE CO2. Graines HEK293-FT cellules 1 jour avant la transfection de sorte qu’ils sont confluents à 80% le jour de la transfection.
REMARQUE : Les cellules HEK293FT sont vaguement adhérentes ; par conséquent, lorsqu’ils sont utilisés pour la production de lentivirus, envisagez de les placage sur des flacons de culture recouverts de 0,1% (w/v) de gélatine pour augmenter l’observance. - Effectuez des transfections le matin. Ajoutez le vecteur de transfert, le mélange d’emballage et le réactif transfection dans les supports compatibles transfection prédémés (p. ex., Opti-MEM). Mélanger en inversant le tube 10-15x. Incuber pendant 5 min à RT. Se référer au tableau 3 pour les volumes exacts.
- Ajouter le réactif transfection comme suggéré par le fabricant. Mélanger par vortexing rapide. Incubate pendant 30 min à RT.
- Aspirer très soigneusement le milieu dépensé. Ajoutez des supports compatibles transfection à la plaque.
- Ajouter le réactif transfection /COMPLEXEs d’ADN tomber sur le côté de la plaque et se propager lentement à travers la plaque en tourbillonnant très doucement.
- Incuber à 37 oC pour 3-5 h et remplacer le milieu par le milieu D10. Incuber toute la nuit.
- Le lendemain matin, remplacez le milieu par un milieu D10 frais. Incuber toute la nuit.
- Le lendemain en fin d’après-midi, recueillir le supernatant viral. Filtrer à l’appel avec un filtre de 0,45 m avec une liaison à faible teneur en protéines. Optionnellement, ajouter frais D10 moyen, incuber toute la nuit et se rappeler le supernatant le lendemain.
- Les supernatants de virus sont stables à 4 oC pendant seulement quelques jours. Conserver à -80 oC pour un stockage à long terme.
REMARQUE : Pour générer une préparation lentivirale très concentrée, qui pourrait être souhaitable pour la transduction de cellules difficiles à transduire, les supernatants peuvent également être concentrés par centrifugation à 6 000 x g pendant la nuit à 4 oC. Marquez la granule virale translucide avec un stylo résistant à l’éthanol et jetez le supernatant. Réutilisez la granule dans 1/100e du volume d’origine pour une augmentation de 100x de la concentration.
- Cellules de culture HEK293-FT dans les médias DMEM/10% FBS à 37 oC et 5% DE CO2. Graines HEK293-FT cellules 1 jour avant la transfection de sorte qu’ils sont confluents à 80% le jour de la transfection.
- Transduire les cellules avec des lentivirus.
- Plaque 1 x 106 cellules par puits dans une plaque de 6 puits avec 3 ml de supports de culture appropriés. Certaines cellules sont plus facilement transduites que d’autres. Pour les cellules faciles à transduire (p. ex., les cellules HEK), ajoutez directement le lentivirus aux cellules. Pour les cellules difficiles à transduire, il pourrait être nécessaire de suivre un protocole de spinoculation tel que décrit ci-dessous.
- Aliquot 2 mL de 2-5 x 106 cellules/mL dans un tube conique de 15 mL.
- Ajouter le lentivirus avec 8 g/mL de bromure d’héraadimethrine et incuber à RT pendant 30 min.
- Centrifugeuse pour 100 min à 800 x g à 32 oC. Puis a resuspendé les cellules dans le même support et ajouter la suspension cellulaire dans les flacons de culture appropriés avec des supports appropriés.
- Autoriser les transductions pendant au moins 24 h. Ensuite, retirez le support contenant le virus et ajoutez un milieu frais.
- Après un autre 24 h, changer les médias à celui qui est complété par les antibiotiques appropriés. La construction Cas9 contient une cassette de résistance blasticidin pour la sélection.
REMARQUE : La quantité de blasticidin doit être optimisée pour chaque lignée cellulaire en effectuant une courbe de mise à mort de réponse à la dose. Une concentration de blasticidine entre 2,5-50 g/mL devrait tuer la plupart des lignées cellulaires non traduites dans les 10 jours suivant la transduction.
- Plaque 1 x 106 cellules par puits dans une plaque de 6 puits avec 3 ml de supports de culture appropriés. Certaines cellules sont plus facilement transduites que d’autres. Pour les cellules faciles à transduire (p. ex., les cellules HEK), ajoutez directement le lentivirus aux cellules. Pour les cellules difficiles à transduire, il pourrait être nécessaire de suivre un protocole de spinoculation tel que décrit ci-dessous.
- Effectuer la sélection jusqu’à ce que toutes les cellules de la plaque de contrôle (c.-à-d. les cellules non traduites qui ont été traitées avec la même concentration d’antibiotiques de sélection) soient tuées.
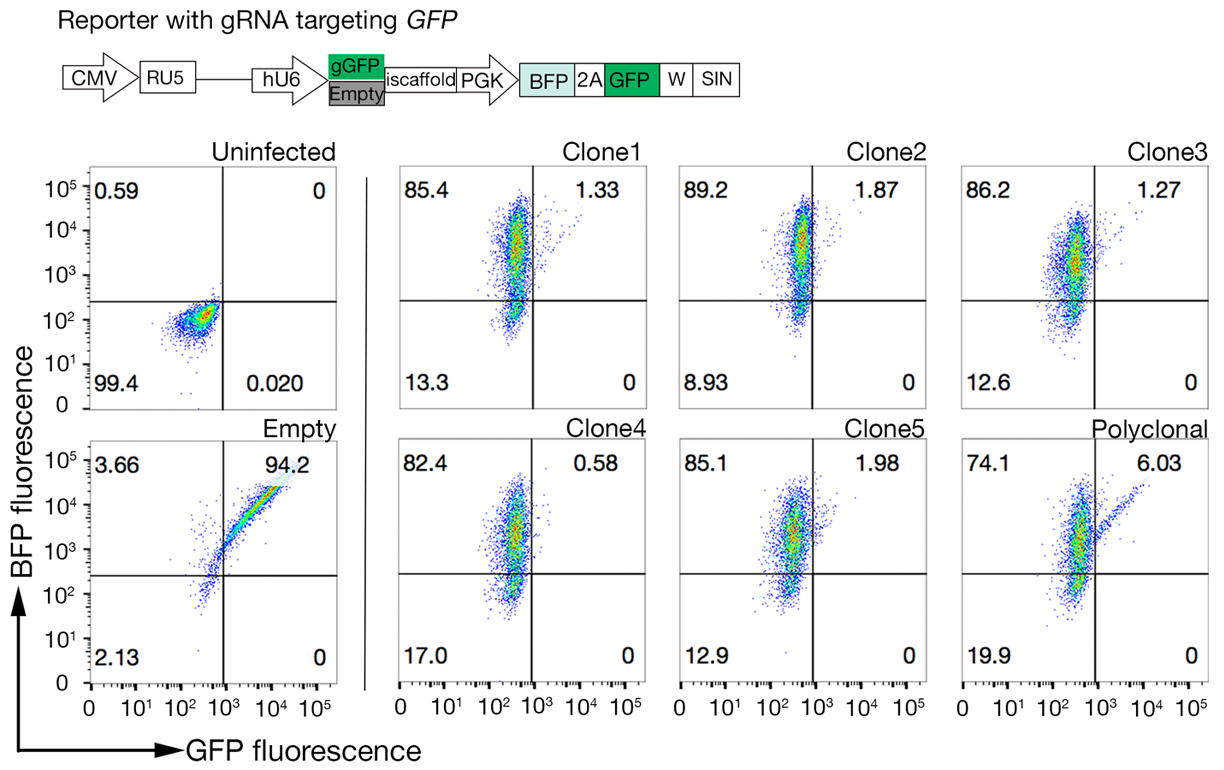
6. Sélection de clones à forte activité Cas9
REMARQUE : Polyclonal Cas9 peut être utilisé pour effectuer avec succès des écrans génétiques ; cependant, la sélection d’un clone avec une activité Cas9 élevée améliore les résultats de dépistage18.
- Utilisez la dilution limitante ou le tri unicellulaire des cellules individuelles résistantes au blasticidine dans des puits de trois 96 plaques de puits contenant des supports de culture complétés par de la blasticidine. Les clones commenceront à émerger entre 2-4 semaines. Sélectionnez 10-20 clones et étendre dans 6 plaques de puits.
- Assay les clones pour l’activité Cas9 en utilisant le système rapide d’évaluation GFP-BFP (protéine fluorescente fluorescente verte-bleu) système, qui utilise un système exogène de knock-out génétique dans lequel les cellules sont transductées soit avec une construction exprimant GFP avec un GFP gRNA-ciblage ou un ARN vide comme un contrôle18.
- plasmides de reporter d’ordre : plasmid de GFP-BFP, plasmid de commande-BFP (tableau 1).
- Produisez le lentivirus pour le plasmide GFP-BFP et le plasmide Control-BFP à l’aide du protocole de production de lentivirus décrit à la section 5.1.
- Transduire chaque clone de ligne cellulaire Cas9-exprimant avec le lentivirus codant le système GFP-BFP et Control-BFP séparément. Suivez le protocole à la section 5.2.
- Après 3 jours de transduction, examinez la fluorescence GFP-BFP de chaque clone à l’aide de la cytométrie du débit. Utilisez 488 nm laser et 405 nm laser pour détecter respectivement GFP et BFP.
- Quantifier l’activité Cas9 dans chaque clone en examinant le rapport de BFP uniquement aux cellules positives GFP-BFP-double. Activité élevée Cas9 cellules devraient idéalement avoir l’efficacité de KO GFP de 95%(figure 4).
7. Génération de la bibliothèque DE dépistage CRISPR-Cas9 à l’échelle du génome
- Pour le criblage à l’échelle du génome à l’aide de la bibliothèque V1humaine 18, commandez la bibliothèque à l’échelle du génome (se référer au tableau 1) et préparez la bibliothèque plasmide à partir du coup bactérien à l’aide du protocole fourni dans le cadre des « Protocoles pour la réplication de la bibliothèque » dans le manuel du fabricant.
- Utilisez la préparation plasmide de bibliothèque à l’échelle du génome pour produire une bibliothèque lentiviral codant des gARN pour la perturbation ciblée des gènes humains à l’aide du protocole de production de lentivirus décrit à la section 5.1.
REMARQUE : Une bonne pratique consiste à produire un seul lot de préparation lentivirale optimisée pour la transduction afin d’améliorer la cohérence expérimentale. - Utilisez le protocole de transduction à la section 5.2 pour effectuer des transductions de test à petite échelle afin de déterminer la quantité requise de virus pour chaque lignée cellulaire pour atteindre une transduction de 30 %. Utilisez la cytométrie des débits pour évaluer la fluorescence BFP comme indicateur de l’efficacité de la transduction.
- Pour transduire les cellules HEK293, il suffit d’ajouter la préparation lentivirale prédéterminée à 30-50 x 106 cellules cultivées dans des supports de croissance normales pour 4 h. Ensuite, retirez les médias par le lentivirus et remplacez-les par de nouveaux supports de croissance.
- Pour d’autres lignées cellulaires, utilisez le protocole de spinoculation à la section 5.2.1 mais à plus grande échelle, de sorte qu’un total de 30-50 x 106 cellules sont transduits. Pour cela, l’aliquot 2 mL de 5 x 106 cellules/mL dans un tube conique de 15 ml et procéder comme indiqué.
- Pour les lignées cellulaires adhérentes, sélectionnez les cellules transduites en ajoutant la puromycine 24 h après la transduction.
REMARQUE : Optimisez les concentrations de puromycine en exécutant une courbe de mise à mort de réponse à la dose. Normalement, les concentrations entre 1-10 g/mL devraient tuer les cellules non traduites dans un délai de 3 à 5 jours. Évitez d’utiliser des concentrations plus élevées de puromycine, car cela peut augmenter les chances de sélection des cellules qui ont été transduites par plus de onesingle guide ARN (sgRNA). - Pour les cellules de suspension, récolter des cellules transduites (c.-à-d. BFP positives) 3 jours après la transmission à l’aide d’un trieur cellulaire et générer des bibliothèques qui contiennent au moins 10 x 106 cellules. Une fois sélectionnés à l’aide de BFP, développer les cellules dans les médias complétés par une quantité appropriée de puromycine.
REMARQUE : Évitez les sélections uniquement avec la puromycine pour les lignées cellulaires de suspension, car il est difficile d’enlever les cellules mortes et les débris des cultures de cellules de suspension qui peuvent interférer avec le tri cellulaire. - Bibliothèque mutante de culture pendant 9-16 jours posttransduction avec passage régulier tous les 2-3 jours.
8. Dépistage génétique de la liaison de surface cellulaire
- Pellet la bibliothèque de cellules mutantes à 200 x g pour 5 min et résutilisez les cellules dans PBS.
- Diviser les cellules en deux tubes coniques de 15 ml avec au moins 50 x 106 cellules dans chaque tube.
- Tourner un tube conique à 200 x g pendant 5 minutes, retirer le supernatant et congeler la pastille cellulaire à -20 oC. Il s’agit de la population témoin et sera traitée plus tard.
- Réutilisez la pastille dans l’autre tube dans 10 ml de PBS/1% BSA. Réserver 100 L de cellules comme un contrôle négatif sur une plaque de puits 96.
- Ajouter la protéine recombinante préconjugée appropriée à la suspension cellulaire dans le tube conique et les protéines de contrôle négatifs à la plaque de puits 96.
- Effectuer la coloration cellulaire pendant au moins 1 h à 4 oC sur un rotor de banc avec rotation douce (6 tr/min).
- Pellet les cellules à 200 x g pendant 5 min, enlever le supernatant. Effectuez deux étapes de lavage, puis réutilisez les cellules dans 5 ml de PBS.
- Filtrer les cellules par une passoire cellulaire de 30 m pour enlever les grappes cellulaires. Analyser à l’aide d’un trieur de flux.
- Utilisez l’échantillon de contrôle négatif pour les cellules BFPMD/PE.
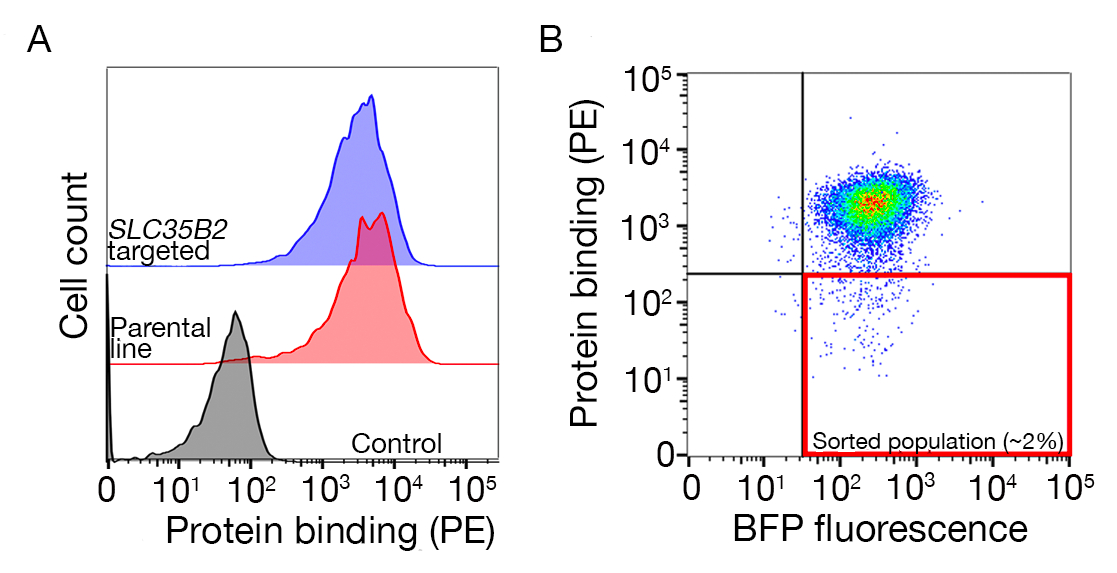
- Trier l’échantillon et recueillir les cellules BFPMD/PE. Les portes de tri dépendront de la liaison des cellules à la protéine mais est normalement 1-5% des échantillons négatifs PE sont recueillis. Un exemple de porte de tri est fourni dans la figure supplémentaire 1.
- Recueillir 500 000 à 1 000 000 cellules de la porte sélectionnée. Compte tenu du faible nombre de cellules, envisagez de prélever les échantillons dans un tube de centrifugation de 1,5 mL pour minimiser les pertes.
- Pelleter les cellules triées en centrifugeant à 500 x g pendant 5 min. Retirez soigneusement le supernatant et jetez. Il est possible de stocker la granule à -20 oC jusqu’à 6 mois.
9. Extraction de l’ADN génomique et premier PCR pour l’enrichissement de l’ARN
- Extraire l’ADN génomique de la population témoin de haute complexité.
- Si la population témoin était gelée à -20 oC, sortez le tube conique et ajoutez PBS. Garder sur la glace pour décongeler la boulette.
- Utilisez un kit commercial (voir Tableau des matériaux)en utilisant les recommandations du fabricant pour extraire l’ADN génomique de 50 x 106 cellules. Ajuster la concentration d’ADN à 1 mg/mL.
- Pour chaque échantillon, configurez un mélange principal pour PCR correspondant à 72 g d’ADN. Aliquot 50 L par puits dans 36 puits d’une plaque de 96 puits PCR. Les séquences d’amorce nécessaires sont répertoriées dans le tableau 4. Utilisez le guide dans les tables 5 et 6.
- Résoudre 5 L du PCR à partir de 6-12 échantillons représentatifs sur un gel d’agarose de 2 % (w/v). Une seule bande claire à 250 bp doit être observée. Si les bandes sont faibles, répétez le PCR pour un cycle supplémentaire de 2-3.
- Utilisez une pipette multicanal pour collecter 5 L de produits PCR de chaque puits (180 l au total) et les mettre en commun dans un réservoir avec 900 l de tampon de liaison à partir d’un kit commercial (voir Tableau des matériaux).
- Purifier les produits PCR à l’aide d’un kit commercial de purification PCR. Elute ADN dans 50 L de tampon d’elution à partir d’un kit commercial (voir Tableau des matériaux) et mesurer la concentration d’ADN.
- Il est peu probable que les échantillons triés pour la perte de phénotype de liaison soient composés d’un grand nombre de clones indépendants. Par conséquent, il n’est pas nécessaire d’effectuer PCR avec 72 g d’ADN. Isoler l’ADN à l’aide d’un kit commercial approprié (voir Tableau des matériaux). Configurez 3-4 réactions DE PCR à l’aide du protocole décrit avant (article 9.1.3) avec 100 ng/L ADN. Si le nombre de cellules triées est inférieur à 100 000, utilisez des lysates cellulaires au lieu de préparations à l’ADN génomique.
- Aliquot environ 10.000 cellules/puits dans une plaque de 96 pcR de puits.
- Pellet les cellules dans la plaque et enlever soigneusement la plupart des supernatants. La pastille ne sera pas visible.
- Ajouter 25 ll d’eau dans chaque puits et chauffer les échantillons à 95 oC pendant 10 min.
- Ajouter 5 ll de 2 mg/mL de proteinase fraîchement diluée K à chaque puits pendant 1 h et incuber à 56 oC. Chauffer ensuite l’échantillon pendant 10 min à 95 oC pour inactiver la proteinase K.
- Utilisez 10 L de mélange de lysate cellulaire par réaction PCR. Les lysates doivent être utilisés dans les 24 h.
10. Deuxième tour de PCR pour le codage à barres index et le séquençage
- Diluer les produits du premier tour PCR à 40 pg/L.
- Configurez un PCR par échantillon (utilisez le guide fourni dans les tableaux 7 et 8). L’utilisation d’une polymérase haute fidélité est importante pour minimiser les erreurs introduites par la polymérase lors de l’amplification de l’ARNm.
- Résoudre 5 L de produits PCR sur un gel agarose de 2 % (w/v). Une seule bande claire à 330 points de base doit être observée.
- Purifier les produits PCR à l’aide de perles paramagnetiques en ajoutant 31,5 L de (0,7x volume total) de perles résinpendées aux produits PCR, en mélangeant bien et en incuber pendant 5 minutes à RT.
- Placer le tube sur une grille magnétique pendant 3 min. Les perles doivent être capturées sur le côté de la plaque et la solution doit être claire. Retirez et jetez soigneusement le supernatant.
- Ajouter 150 l d’éthanol fraîchement préparé à 80 % dans le tube. Incuber pendant 30 s, puis retirer soigneusement et jeter le supernatant.
- Répétez l’étape 13.6, cette fois avec 180 L. Puis sécher l’air les perles pendant 5 min.
- Retirez le tube de l’aimant. Cible d’ADN Elute des perles en 35 L de tampon EB stérile. Incuber pendant 3 minutes, puis remettre le tube dans la grille magnétique pendant 3 minutes.
- Transférer environ 30 L du supernatant contenant les produits PCR enlisés dans un tube propre.
- Séquencez les échantillons sur une plate-forme de séquençage de nouvelle génération. Pour la bibliothèque d’ARN De HumanV1, utilisez l’amorce personnalisée énumérée dans le tableau 4 pour séquencer 19 bp.
11. Analyse bioinformatique pour identifier le récepteur et les voies connexes
- Séquences de cartes de la population triée et non triée à la bibliothèque de référence en utilisant la fonction de comptage de MAGeCK. La fonction donnera un fichier de comptage brut(tableau supplémentaire 1).
REMARQUE : Des instructions détaillées sur l’installation de MAGeCK et l’utilisation de différentes fonctions au sein de MAGeCK sont décrites dans un protocole publié précédemment par Wang et coll.20. - Vérifiez la norme technique de la bibliothèque de contrôle utilisée dans l’écran.
- La médiane normalise le nombre brut et utilise le paquet ggplot2 en R21 ou un logiciel équivalent pour tracer une parcelle empirique de la fonction de densité cumulative des comptes dans les échantillons plasmides et de contrôle des échantillons non triés(figure 5A).
- Exécuter la fonction de test de MAGeCK en utilisant les comptes de la population plasmide comme « contrôle » et les comptes des échantillons de contrôle non triés comme l’échantillon « test ». La fonction est donnera un fichier de résumé de gène(tableau supplémentaire 1).
- Ouvrez le fichier de résumé génétique et dessinez la distribution des changements de journal (colonneneg-lfc) pour les gènes essentiels et non essentiels précédemment classés22 (figure 5B).
- Sélectionnez des gènes considérablement appauvris(neg-fdr et 0,05) et effectuez l’analyse de l’enrichissement de la voie à l’aide de l’emballage23 de l’enrichissement de l’enrichissementde l’enrichissement de l’enrichissement de la voie ou de tout emballage équivalent d’enrichissement de voie en R(figure 5C).
- Exécutez la fonction de test de MAGeCK avec paramètre par défaut. Utilisez les nombres bruts de l’échantillon de contrôle non triés comme «contrôle» et compte de l’échantillon trié comme «traitement» lors de l’exécution de l’analyse.
- Ouvrez le fichier de résumé de gènes généré par MAGeCK et classez la colonne de rang pos dans l’ordre ascendant. Utilisez FDR (colonnede pos-fdr) 'lt; 0.05 comme coupure pour l’identification des coups. Le récepteur est généralement classé haut, souvent en première position.
- Tracez les scores Robust-Ranking-Algorithm (RRA) pour une sélection positive(pos-score)en R ou un logiciel équivalent(figure 5D).
- Sélectionnez les coups génétiques et effectuez l’analyse de l’enrichissement de la voie à l’aide de l’emballage d’enrichissement ou de tout paquet équivalent d’enrichissement de voie en R pour identifier les voies enrichies.
Résultats
Les données de séquençage de deux écrans à élimination directe représentatifs à l’échelle du génome pour l’identification du partenaire contraignant de l’humain TNFSF9 et P. falciparum RH5 effectués respectivement dans les cellules NCI-SNU-1 et HEK293 sont fournies(tableau supplémentaire 1). Le comportement contraignant de RH5 a été affecté par le sulfate de chaparan et son récepteur connu BSG24 (figure 3C), tandis que TNFRSF9 spécifiquement lié à son récepteur connu TNFSF9 et n’a pas perdu la liaison à la préincubation avec l’héparine soluble. La protéine 3 de la figure 3B représente le TNFRSF9.
Pour les deux lignées cellulaires, la distribution des ARNN dans la bibliothèque de mutants témoins après 3 jours (9, 14 et 16 jours posttransduction) sont également fournies(tableau supplémentaire 1). La distribution de l’ARN a révélé que la complexité de la bibliothèque a été maintenue tout au long de l’expérience(figure 5A). L’écran génétique pour l’identification du ligand pour TNFSF9 a été effectué le jour 14 posttransduction, alors que celui pour RH5 a été effectué jour 9 posttransduction. La qualité technique des écrans a été évaluée en examinant la distribution des changements croisés observés des ARNmaux ciblant un ensemble de référence de gènes non essentiels par rapport à la distribution pour l’ensemble de référence des gènes essentiels22 (figure 5B). De plus, l’enrichissement au niveau des voies a également révélé que les voies essentielles attendues ont été identifiées et considérablement enrichies dans la population « décrochage » lorsqu’elles comparent l’échantillon témoin à la bibliothèque plasmide originale. Un exemple avec l’échantillon du jour 14 NCI-SNU-1 est représenté dans la figure 5C.
La distribution des ARN gRN dans la population témoin par rapport à la population triée à l’aide de la fonction de test de MAGeCK (voir tableau supplémentaire 1 pour la sortie sommaire du gène de MAGeCK) a été utilisée pour identifier le récepteur des écrans phénotypiques. Le score modifié de RRA rapporté par MAGeCK dans l’analyse de gene-niveau est tracé contre les gènes classés par les valeurs de p. Le score de la RRA dans MAGeCK fournit une mesure dans laquelle les ARNN sont classés constamment plus élevés que prévu. Dans l’écran de TNFRSF9, le meilleur succès a été TNFSF9, qui est un partenaire de liaison connu de TNFRSF9(figure 5D). En outre, un certain nombre de gènes liés à la voie TP53 ont également été identifiés. Dans le cas de RH5, en plus du récepteur connu (BSG) et du gène requis pour la production des GAF sulfés(SLC35B2), un gène supplémentaire (SLC16A1) a également été identifié(figure 5E). SLC16A1 est un chaperon nécessaire pour le trafic BSG à la surface des cellules25. Ensemble, ces résultats démontrent la capacité de l’écran à identifier les récepteurs qui interagissent directement et les composants cellulaires nécessaires pour que ce récepteur soit exprimé à la surface des cellules sous une forme fonctionnelle.

Figure 1 : Aperçu de l’approche de dépistage génétique pour identifier les récepteurs de surface cellulaire. Cet essai se compose de trois étapes majeures : Premièrement, les protéines recombinantes représentant l’ectodomain des récepteurs de surface cellulaire sont exprimées dans une lignée cellulaire qui peut ajouter des modifications posttranslationnelles structurellement critiques telles que les cellules HEK293. Les ectodomains de protéine monomérique sont oligomerized par la conjugaison à streptavidin-PE pour augmenter leur avidité de liaison. Deuxièmement, ces sondes avides sont utilisées dans les essais de liaison cellulaire où la coloration lumineuse sur les lignées cellulaires indiquée par un changement important dans la fluorescence PE (en vert) par rapport à une protéine de contrôle négatif (en noir) démontre la présence d’un partenaire de liaison de surface cellulaire. Troisièmement, les lignées cellulaires cas9-exprimant les récepteurs sont sélectionnées et le criblage à l’échelle du génome à l’aide de gARN ciblant la grande majorité des gènes de codage des protéines est effectué. Tout en générant des bibliothèques mutantes, il est courant d’utiliser une efficacité de transduction de 30 %, qui est basée sur la probabilité de distribution de Poisson qui garantit que chaque cellule reçoit un seul ARN de sorte que le phénotype résultant est attribué à un KO spécifique. Le marqueur BFP exprimé par les cellules transduites est utilisé pour sélectionner les cellules contenant des ARNr à l’aide de FACS. Les écrans phénotypiques sont effectués entre 9-16 jours posttransduction. Le jour de l’écran, la population totale de cellules mutantes est divisée en deux. Une moitié est conservée comme population témoin et l’autre moitié est sélectionnée pour la liaison de protéine recombinante. Les cellules de la bibliothèque mutante qui ne sont plus en mesure de lier la protéine recombinante sont triées à l’aide de FACS et l’enrichissement des ARN dans la population triée par rapport à la population témoin est utilisé pour identifier les gènes nécessaires à la liaison de surface cellulaire de la sonde avide étiquetée. Les étapes du protocole qui nécessitent beaucoup de temps sont indiquées. Ce chiffre a été modifié à partir de Sharma et al.19. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Établir les ratios de protéines biotinylateds à la streptavidine-PE à l’aide d’une méthode à base d’ELISA. Un exemple de stratégie de conjugaison-PE streptavidin-PE utilisé pour générer une sonde avide à partir d’une protéine monoméane biotinylated. Une série de dilution des monmers biotinylated a été incubée contre une concentration fixe de streptavidin. La dilution minimale à laquelle aucun excès de monomères biotinylated ne peut être détectée a été déterminée par ELISA. ELISA a été exécuté avec ou sans préincubation d’une gamme de dilutions de protéine avec 10 ng de streptavidin-PE. En présence de streptavidin-PE, la dilution minimale à laquelle aucun signal n’a été identifié (noir encerclé) et la quantité de protéines nécessaires à la saturation ont été calculées pour générer une solution de stock de 10x avec 4 g/mL streptavidin-PE. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Liaison représentative des protéines aux lignées cellulaires. (A) La liaison protéique aux lignées cellulaires a eu une augmentation claire de la fluorescence cellulaire-associée par rapport à l’échantillon témoin. Le traitement thermique (80 oC pendant 10 min) de protéines recombinantes a abrogé toute liaison vers un contrôle négatif, ce qui démontre que le comportement contraignant dépendait de protéines correctement pliées. (B) Différentes classes de comportement de liaison de protéine aux surfaces cellulaires ; dépendance vis-à-vis des GAG. De gauche à droite, les protéines peuvent être classées en trois types : Protein type 1 seulement adsorbs à HS. Ces protéines perdent leur liaison après la préincubation avec des concentrations d’héparine supérieures à 0,2 mg/mL. Le type 2 des protéines se lie à HS en plus d’un récepteur spécifique. Ces protéines perdent la liaison partielle dans les expériences de préblocage. Le type de protéine 3 ne lie pas HS. Ces protéines ne perdent pas de liaison par rapport aux lignées parentales. (C) Un exemple d’une protéine (c.-à-d., RH5) qui se lie à HS et un récepteur spécifique d’une manière additive. Cibler soit le récepteur (c’est-à-dire BSG) ou les enzymes nécessaires à la synthèse HS (p. ex., SLC35B2, EXTL3) ne réduit que partiellement la liaison du RH5 aux cellules par rapport aux témoins. Les lignes polyclonal transduites peuvent être utilisées dans de telles expériences pour établir un comportement contraignant. Ce chiffre a été modifié à partir de Sharma et al.19. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Sélection des lignées cellulaires clonales à forte activité Cas9. L’efficacité de l’édition génomique des lignées cellulaires NCI-SNU-1 a été évaluée à l’aide du système de reporter GFP-BFP, dans lequel les lignées cellulaires étaient transduites avec des virus avec un plasmide de ciblage de l’ARNC codé GFP ou sans (c.-à-d. « vide »). Un schéma est représenté. La cytométrie de débit a été employée pour tester l’expression de BFP et de GFP après transduction et comparée au contrôle non infecté. L’expression GFP a été utilisée comme proxy pour l’activité Cas9, tandis que l’expression BFP a marqué les cellules transduites. Le profil des cellules infectées non infectées et vides ressemblait à tous les clones. Les profils représentatifs sont représentés dans le panneau gauche. Les cinq clones de la lignée cellulaire NCI-SNU-1 ont montré une perte plus élevée de GFP par rapport à la ligne polyclonal (panneau droit), avec le clone 4 montrant la plus haute efficacité avec la population réfractaire la plus faible. Ce chiffre a été modifié à partir de Sharma et al.19. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Les résultats représentatifs des écrans génétiques pour l’identification des partenaires de liaison de surface cellulaire. (A) Les parcelles cumulatives de fonction de distribution comparant l’abondance d’ARN dans la bibliothèque plasmide aux bibliothèques mutantes des cellules HEK-293-E et NCI-SNU-1 le jour 9, 14 et 16 jours posttransduction. Pour un nombre donné, la fonction de densité cumulative indique le pourcentage de points de données qui étaient inférieurs à ce seuil. Le petit déplacement de la population de cellules mutantes par rapport à la population plasmide originale représente l’épuisement dans un sous-ensemble de gARN par rapport à la bibliothèque plasmide. (B) Distribution des changements de pliage des gènes qui ont été précédemment classés comme étant essentiels (rouge) ou non essentiels (noir) dans les lignées cellulaires HEK293 et NCI-SNU-1. La distribution des changements de plis pour les gènes non essentiels centrés à 0, tandis que pour les gènes essentiels déplacé vers la gauche vers des changements négatifs de pli. (C) Voies significativement enrichies dans les gènes appauvris dans NCI-SNU-1 population de contrôle mutant 14 jours posttransduction. Des voies essentielles connues attendues de cellules ont été identifiées. (D) Robust Rank Algorithm (RRA)-score pour les gènes qui ont été enrichis dans les cellules triées qui avaient perdu la capacité de lier la sonde TNFRSF9. Les gènes ont été classés selon le RRA-score. Le partenaire d’interaction connu TNFSF9 et les gènes liés à la voie TP53 (étiquetés en rouge) ont été identifiés dans l’écran. (E) Scores RRA classés rang pour les gènes identifiés à partir de l’analyse d’enrichissement de l’ARNm requise pour la liaison RH5 aux cellules HEK293 (panneau gauche). SLC35B2 et SLC16A1 ont été identifiés dans un seuil de faux-découverte (FDR) de 5%. Deux autres gènes de la voie de la biosynthèse HS (c.-à-d. EXTL3 et NDST1)ont été identifiés dans le FDR de 25 %. Schématographe représentant la voie générale de biosynthèse GAG avec les gènes pertinents cartographiés aux étapes correspondantes (panneau 2). Les gènes requis pour l’engagement de biogenèse de sulfate de chondroïtine (c.-à-d. CSGALNACT1/2) n’ont pas été identifiés dans l’écran. Ce chiffre a été modifié à partir de Sharma et al.19. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
| Nom Plasmid | Plasmide # | Utiliser |
| Construction d’expression protéique : CD200RCD4d3-4-bio-linker-his | Addgene: 36153 | Production de protéines recombinantes avec CD4d3-4, biotine et 6-ses étiquettes. |
| pMD2.G (en) | Addgene: 12259 | enveloppe VSV-G exprimant plasmide; production de lentivirus |
| psPAX2 (en) | Addgene: 12260 | plasmide d’emballage lentiviral, production de lentivirus |
| Cas9-construction: pKLV2-EF1a-Cas9Bsd-W | Addgene: 68343 | Production de la ligne Cas9 qui exprime la constitutive |
| gRNA expression build: pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W | Addgene: 67974 | Vecteur d’expression CRISPR gRNA avec un échafaudage amélioré et des marqueurs puro/BFP |
| Bibliothèque Knockout CRISPR à l’échelle du génome humain | Addgene: 67989 | Une bibliothèque d’ARN contre 18 010 gènes humains, conçue pour être utilisée dans le lentivirus. |
| GFP-BFP construction: pKLV2-U6gRNA5(gGFP)-PGKBFP2AGFP-W | Addgene: 67980 | Journaliste d’activité Cas9 avec BFP et GFP. |
| Construction vide: pKLV2-U6gRNA5(vide)-PGKBFP2AGFP-W | Addgene: 67979 | Journaliste d’activité Cas9 (contrôle) avec BFP et GFP. |
Tableau 1 : Plasmides utilisés dans cette approche.
| Nom tampon | Composants |
| HBS (10X) | 1,5 M NaCl et 200 mM HEPES dans l’eau MiliQ, ajuster à pH 7.4 |
| PBS (10X) | 80 g De NaCl, 2 g KCl, 14,4 g Na2HPO4 et 2,4 g KH2PO4 dans l’eau MiliQ, ajuster au pH 7,4 |
| Tampon de phosphate de sodium (80mm stock) | 7,1 g Na2HPO4.2H2O, 5,55 g NaH2PO4, ajuster au pH 7.4 |
| Son tampon de liaison de purification | 20 mM sodium Phosphate Buffer, 0,5 M NaCl et 20 mM Imidazole, ajuster à pH 7,4 |
| Son tampon d’elution de purification | 20 mM sodium Phosphate Buffer, 0.5M NaCl et 400 mM Imidazole, ajuster à pH 7.4 |
| Tampon de dénominamine | 10% d’éthanolamine et 0,5 mM MgCl2 dans l’eau MiliQ, ajuster à pH 9.2: |
| D10 D10 | DMEM, 1% pénicilline-streptomycine (100 unités/mL) et 10% de chaleur inactivée FBS |
Tableau 2 : Tampons requis pour cette étude.
| Composants | Plat de 10 cm | Plaque de 6 puits |
| Cellules 293FT | 70 à 80 % confluents | 70 à 80 % confluents |
| Médias compatibles transfection (Opti-MEM) (étape 5.1.2) | 3 mL | 500 l |
| Médias compatibles transfection (Opti-MEM) (étape 5.1.4) | 5 mL | 2 ml |
| Vecteur de transfert lentiviral | 3 g | 0,5 g |
| psPax2 (voir tableau 1) | 7,4 g | 1,2 g |
| pMD2.G (voir tableau 1) | 1,6 g | 0,25 g |
| PLUS réactif | 12 l | 2 l |
| Lipofectamine LTX | 36 l | 6 ll |
| D10 (étape 7.1.7) | 5 mL | 1,5 mL |
| D10 (étape 7.1.8 et 7.1.10) | 8 ml | 2 ml |
Tableau 3 : Montants et volumes de réactifs pour le mélange d’emballage lentivirus.
Tableau 4 : Séquences d’apprêt pour amplifier l’ARNM et le NGS. S’il vous plaît cliquez ici pour afficher ce fichier (Cliquez à droite pour télécharger).
| Réactif | Volume par réaction | Master mix (x38) |
| Q5 Hot Start High-Fidelity 2x | 25 ll | 950 l |
| Mélange d’apprêt (L1/U1) (10 m chacun) | 1 l | 38 ll |
| ADN génomique (1 mg/mL) | 2 l | 72 l |
| H2O | 22 l | 1100 l |
| Total | 50 l | 1900 l |
Tableau 5 : PCR pour l’amplification des ARN gRN à partir d’échantillons de haute complexité.
| Numéro de cycle | Dénaturer | Recuit | Extension |
| 1 | 98 oC, 30s | ||
| 2-24 | 98 oC, 10s | 61 oC, 15s | 72 oC, 20s |
| 25 | 72 oC, 2 min |
Tableau 6 : Conditions PCR pour le premier PCR.
| Réactif | Volume par réaction |
| KAPA HiFi HotStart ReadyMix | 25 ll |
| Mélange d’apprêt (PE1.0/index primer) (5 m chacun) | 2 l |
| Premier produit PCR (40 pg/L) | 5 ll |
| H2O | 18 l |
| Total | 50 l |
Tableau 7 : PCR pour l’index de marquage des sgARN à partir d’écrans génétiques.
| Numéro de cycle | Dénaturer | Recuit | Extension |
| 1 | 98 oC, 30s | ||
| 2-15 | 98 oC, 10s | 66 oC, 15s | 72 oC, 20s |
| 16 | 72 oC, 5 min |
Tableau 8 : Conditions PCR pour le deuxième PCR.

Figure supplémentaire S1 : Guide pour dessiner des portes pour trier la population non contraignante. (A) Un candidat idéal pour le dépistage de protéine devrait avoir un changement clair de la population liante comparée à la population témoin et la liaison devrait être conservée sur les cellules manquant de machines pour la biosynthèse de HS. Une expérience de blocage d’héparine peut être utilisée à la place d’essais sur les lignées cellulaires ciblées SLC35B2. (B) Des cellules dépourvues de la coloration de surface de l’ectodomain de protéine mais exprimant la fluorescence de BFP de la transduction lentiviral ont été rassemblées. Les cellules affichées proviennent d’un écran pour l’identification du récepteur pour GABBR222. Ce chiffre a été modifié à partir de Sharma et al.19. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

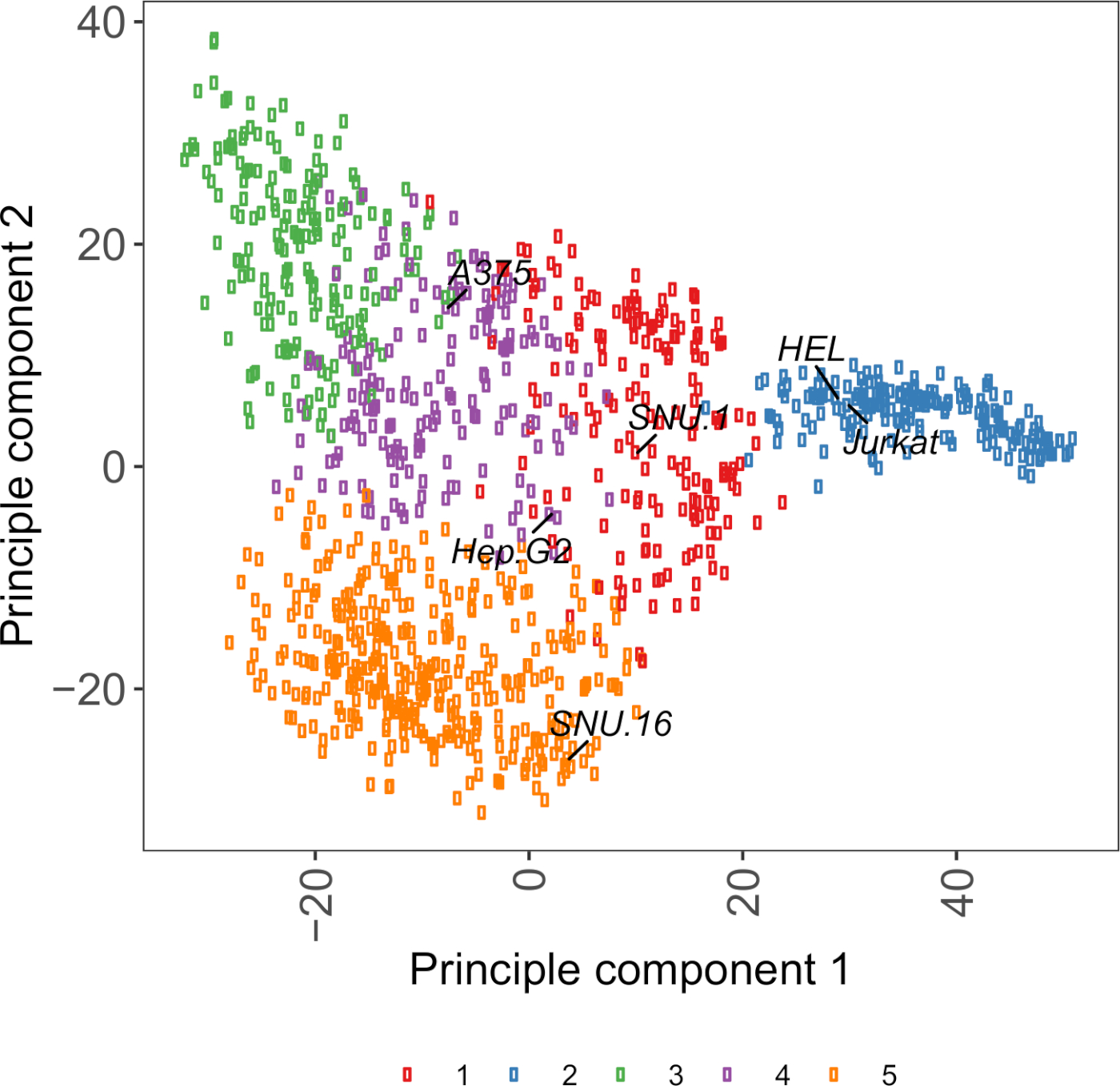
Figure supplémentaire S2 : Cellule glycoprotéine de surface transcriptomics base de parcelle de PCA à l’aide des données RNA-seq de plus de 1.000 lignées de cellules cancéreuses. Les lignées cellulaires de Cell Model Passport27 ont été regroupées à l’aide de la grappe de K-moyens selon les valeurs FPKM de 1 500 glycoprotéines de surface cellulaire. Les lignées cellulaires représentatives de chaque cluster sont étiquetées. Le cluster 5 était entièrement composé de lignées cellulaires d’origine hématopoïétique (voir également tableau supplémentaire 2). S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

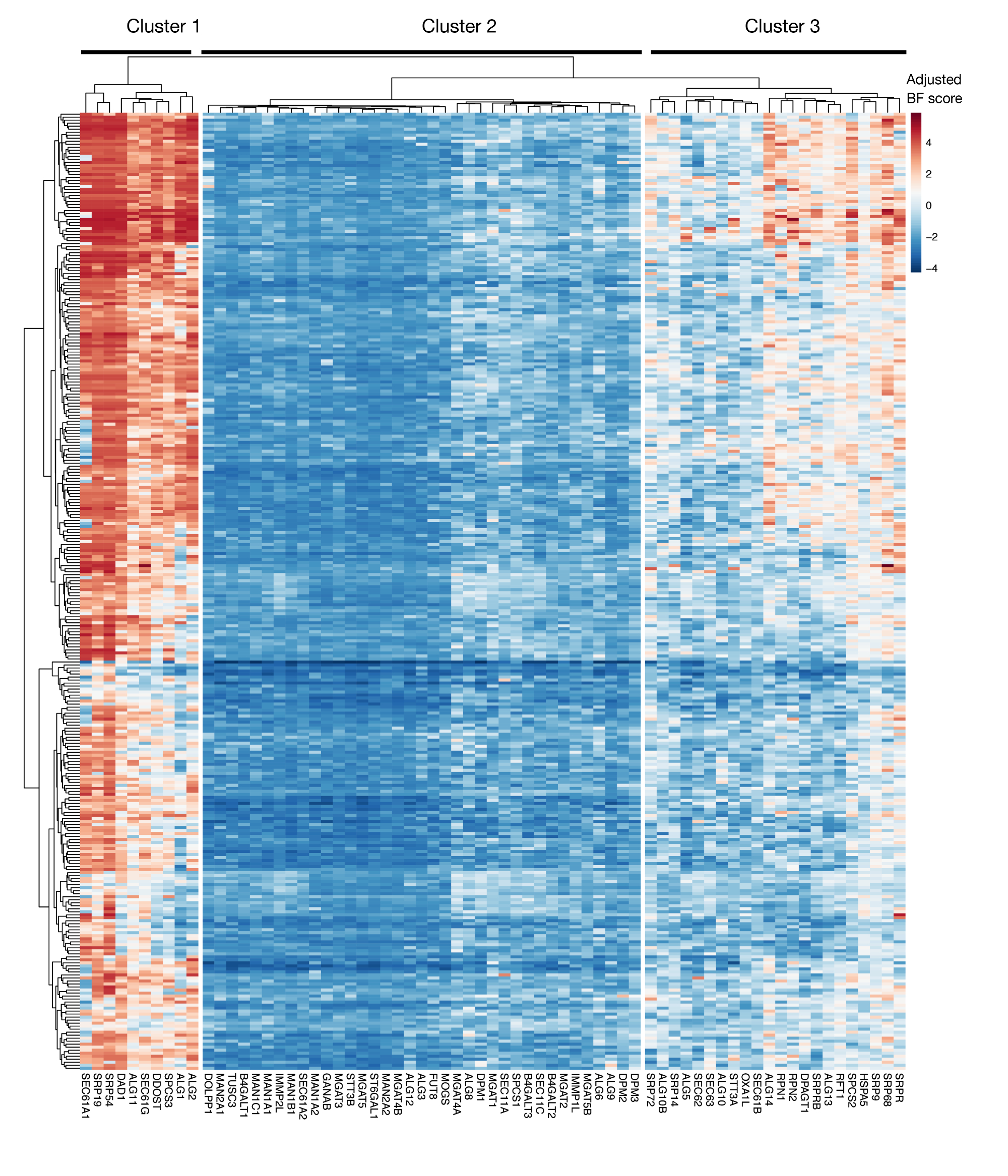
Figure supplémentaire S3 : Scores essentiels pour l’exportation de protéines d’annotation de KEGG et les gènes de glycosylation liés au N des scores de projet. Les scores d’essentialité ajustées de Bayes pour 330 lignées cellulaires (colonnes, non étiquetées) sont tracés pour les gènes d’exportation de protéines et de la voie de glycosylation liée au N (axe X). Les scores supérieurs à 0 représentent un épuisement significatif de la population mutante par rapport à la bibliothèque plasmide originale. Les gènes peuvent être divisés en trois amas distincts qui représentent différents niveaux d’essentialité dans les lignées cellulaires. Ce regroupement peut être utilisé pour décider du jour du tri. Si l’écran est effectué à un point de retard (jour 16), il est possible que les gènes qui sont connus pour être essentiels pour les cellules (clusters 1 et 3) ne seront pas identifiés. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Tableau supplémentaire 1 : Les fichiers de comptage brut et le logiciel MAGeCK ont généré gene_summary fichiers liés aux écrans génétiques représentatifs. S’il vous plaît cliquez ici pour afficher ce fichier (Cliquez à droite pour télécharger).
Tableau supplémentaire 2 : Regroupement des lignées cellulaires selon l’expression des récepteurs de surface cellulaire. S’il vous plaît cliquez ici pour afficher ce fichier (Cliquez à droite pour télécharger).
Discussion
Une stratégie de dépistage basée sur CRISPR pour identifier les gènes codant des composants cellulaires impliqués dans la reconnaissance cellulaire est décrite. Une approche similaire utilisant l’activation CRISPR fournit également une alternative génétique pour identifier les récepteurs directement interagissant des protéines recombinantes sans avoir besoin de générer de grandes bibliothèques de protéines26. Cependant, un avantage majeur de cette approche est qu’elle s’applique aux interactions négociées par des molécules de surface affichées localement sur la cellule et ne dépend pas de la surexpression des récepteurs, ce qui peut influencer l’avidité contraignante du récepteur. Contrairement à d’autres méthodes, cette technique ne fait donc aucune hypothèse concernant la nature biochimique ou la biologie cellulaire des récepteurs et offre l’occasion d’étudier les interactions médiatisées par des protéines qui sont normalement difficiles à étudier à l’aide d’approches biochimiques, telles que les très grandes protéines, ou celles qui traversent la membrane plusieurs fois ou forment des complexes avec d’autres protéines, et des molécules autres que des protéines telles que les glycans, les glycolipids et les phosphospholipides. Compte tenu de la nature à l’échelle du génome de la méthode, cette approche a également l’avantage non seulement d’identifier le récepteur, mais aussi d’autres composants cellulaires qui sont nécessaires pour l’événement contraignant, fournissant ainsi un aperçu de la biologie cellulaire du récepteur.
Une des principales limites de cette méthode lors de son utilisation pour identifier le récepteur d’une protéine orpheline est l’exigence initiale d’identifier d’abord une lignée cellulaire qui se lie à la protéine. Ce n’est pas toujours facile et l’identification d’une lignée cellulaire qui affiche un phénotype de liaison qui est également permissif aux écrans génétiques peut être l’étape limite de temps pour le déploiement de cet essai. Certaines lignées cellulaires ont tendance à se lier à plus de protéines que d’autres. Ceci est particulièrement pertinent pour les protéines qui lient HS, parce que ces protéines ont tendance à se lier à n’importe quelle lignée cellulaire qui affiche des chaînes latérales HS, indépendamment du contexte de liaison indigène. En outre, nous avons observé que l’amélioration de la réglementation des syndécans (c’est-à-dire les protéoglycans qui contiennent du HS) dans les lignées cellulaires conduit à une liaison accrue des protéines HS-contraignantes26. Cela pourrait être un facteur à prendre en considération lors de la sélection de la ligne cellulaire pour le dépistage. Cependant, il est également important de noter que la liaison additive de HS n’interfère pas avec la liaison à un récepteur spécifique. Cela signifie que si la liaison est observée, il est possible qu’elle soit négociée uniquement par HS parce que la liaison négociée par HS dans cet essai est additive plutôt que codépendant19. Dans un tel scénario, l’approche de blocage de l’héparine décrite peut identifier de tels comportements sans avoir besoin d’effectuer un écran génétique complet.
Une ressource utile pour le choix des lignées cellulaires est Cell Model Passport, qui contient de la génomique, de la transcriptomique et de l’information sur l’état de la culture pour 1 000 lignées de cellules cancéreuses27. Selon le contexte biologique, les cellules peuvent être choisies en fonction de leurs profils d’expression. Pour faciliter la sélection des lignées cellulaires, nous avons regroupé 1 000 lignées cellulaires dans Cell Model Passport selon l’expression de 1 500 glycoprotéines de surface des cellules humaines préannotées28 (figure supplémentaire 2; des informations de cluster pour chaque lignée cellulaire ainsi que des conditions de croissance sont fournies dans le tableau supplémentaire 2). Lors de l’essai de la liaison d’une protéine avec une fonction inconnue, il est utile de sélectionner un panel de lignées cellulaires représentatives de chaque cluster pour augmenter les chances de couvrir un large éventail de récepteurs. Étant donné le choix, il est recommandé de choisir des lignées cellulaires faciles à culture et faciles à transduire. Comme ces lignées cellulaires seront utilisées dans le criblage à l’échelle du génome, il est préférable qu’elles puissent être cultivées facilement en grandes quantités et sont permissives à la transduction lentivirale, parce qu’il s’agit de la méthode la plus couramment disponible pour la livraison du sgRNA pour le dépistage génétique à base de CRISPR dans les étapes ultérieures.
En général, les sélections de phénotypes sont effectuées en une seule sorte. Cependant, cela est déterminé par la luminosité de la population de cellules tachées par rapport au contrôle. Des rondes itératives de sélections pourraient être adoptées pour des scénarios dans lesquels le rapport signal-bruit du phénotype désiré est faible, ou lorsque le but de l’écran est d’identifier les mutants qui ont des phénotypes forts. Lors de l’utilisation d’une approche de sélection itérative pour les écrans à base de FACS, il est important de considérer que le processus de tri peut causer la mort cellulaire, principalement en raison de la force pure du trieur. Ainsi, toutes les cellules collectées ne seront pas représentées dans la prochaine série de tri.
La complexité de la bibliothèque est un facteur très important dans l’exécution des écrans génétiques réussis, en particulier pour les écrans de sélection négatifs parce que l’ampleur de l’épuisement dans ceux-ci ne peut être déterminée qu’en comparant les résultats à ce qui était présent dans la bibliothèque de départ. Pour les écrans de sélection négatifs, il est courant de maintenir des bibliothèques de 500 à 1 000 x complexité. Les écrans de sélection positifs, cependant, sont plus robustes pour les tailles de bibliothèque, parce que dans ces écrans seulement un petit nombre de mutants sont censés être sélectionnés pour un phénotype particulier. Par conséquent, dans l’écran de sélection positif décrit ici, la taille de la bibliothèque peut être réduite à la complexité 50-100x sans compromettre la qualité de l’écran. En outre, dans ces écrans, il est également possible d’utiliser une bibliothèque de contrôle pour une ligne cellulaire donnée un jour donné comme un « contrôle général » pour tous les échantillons triés le jour pour cette ligne cellulaire donnée. Cela réduira le nombre de bibliothèques de contrôle qui doivent être produites et séquencées.
Une autre considération importante pour l’utilisation de cette approche est les limites des écrans de perte de fonction dans l’identification des gènes qui sont essentiels pour la croissance des cellules in vitro. Le moment des écrans est crucial à cet égard, car plus les cellules mutantes sont maintenues dans la culture, plus la probabilité que les cellules avec des mutations dans les gènes essentiels deviennent non viables et ne sont plus représentés dans la bibliothèque mutante. Les récents tests génétiques effectués dans le cadre de l’initiative Project Score dans plus de 300 lignées cellulaires montrent que plusieurs gènes de la sécrétion de protéines annotée keGG et de la voie de la N-glycosylation sont souvent identifiés comme étant essentiels pour un certain nombre de lignées cellulaires(figure supplémentaire 3)29. Cela peut être pris en considération lors de la conception des écrans si l’effet des gènes nécessaires à la prolifération et la viabilité doit être étudié dans le cadre du processus de reconnaissance cellulaire. Dans ce cas, l’exécution d’écrans à un point d’heure précoce (p. ex., posttransduction du jour 9) serait généralement appropriée. Toutefois, si l’approche est utilisée pour identifier quelques cibles ayant de forts effets de taille plutôt que des voies cellulaires générales, il pourrait être approprié d’effectuer des écrans à un moment ultérieur (p. ex., posttransduction du jour 15-16).
Les résultats du dépistage sont très solides; dans huit écrans recombinés de liaison de protéine exécutés dans le passé, le récepteur de surface de cellules a été le coup supérieur dans chaque cas19. Lorsque vous utilisez cette approche pour identifier le partenaire d’interaction, il faut donc s’attendre à ce que le récepteur et les facteurs contribuant à sa présentation à la surface soient identifiés avec une confiance statistique élevée. Une fois que l’écran est exécuté et qu’un coup est validé à l’aide d’un seul knock-out d’ARN, d’autres suivis peuvent être effectués à l’aide de méthodes biochimiques existantes telles que l’AVEXIS4 et la liaison saturable directe des protéines purifiées à l’aide d’une résonance plasmon de surface. L’approche décrite ici s’applique à toutes les protéines pour lesquelles il est possible de générer une sonde recombinante soluble.
En résumé, il s’agit d’une approche à élimination directe CRISPR à l’échelle du génome pour identifier les interactions négociées par les protéines de surface cellulaire. Cette méthode s’applique généralement pour identifier les voies cellulaires requises pour la reconnaissance de surface cellulaire dans un large éventail de contextes biologiques différents, y compris entre les propres cellules d’un organisme (p. ex., la reconnaissance neuronale et immunologique), ainsi qu’entre les cellules hôtes et les protéines pathogènes. Cette méthode offre une alternative génétique aux approches biochimiques conçues pour l’identification des récepteurs, et parce qu’elle ne nécessite aucune hypothèse préalable concernant la nature biochimique ou la biologie cellulaire des récepteurs, elle a un grand potentiel pour faire des découvertes complètement inattendues.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce travail a été soutenu par le numéro de subvention Wellcome Trust 206194 attribué à GJW. Nous remercions l’installation Cytometry Core : Bee Ling Ng, Jennifer Graham, Sam Thompson et Christopher Hall pour l’aide de FACS.
matériels
| Name | Company | Catalog Number | Comments |
| Anti-mouse alkaline phosphatase | Sigma | A4656 | |
| Blasticidin | Chem-Cruz | SC-204655 | |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | 13362 | |
| BSA | Sigma | A9647-100G | |
| Diethanolamine | Sigma | 398179 | |
| DMEM | Gibco | 31966-021 | |
| Dneasy Blood and Tissue kit | Qiagen | 69504 | |
| DynaMag-96 Side Magnet | Invitrogen | 12331D | |
| HEK293T packaging cells | ATCC | CRL-3216 | |
| Heparin | Sigma | H4784-1G | |
| KAPA HiFi HotStart ReadyMix | Kapa | KK2602 | |
| Lipofectamine LTX with PLUS reagent | Invitrogen | 15338100 | |
| MoFlo XDP cell sorter | BD | ||
| Ni2+-NTA agarose beads | Jena Bioscience | AC-501-25 | |
| OPTI-MEM | Life Technologies | 31985-070 | |
| OX-68 antibody | AbD Serotec | MCA1022R | |
| p-nitrophenyl phosphate | Sigma | 1040-506 | |
| PD-10 desalting columns | GE healthcare | 17085101 | |
| Polybrene | Millipore | TR-1003-G | |
| Polypropylene tubes with 5 mL bed volume | Qiagen | 34964 | |
| Proteinase K, recombinant, PCR Grade | Roche | 3115879001 | |
| Puromycin | Gibco | A11138-03 | |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | M0494L | |
| QIAquick PCR purification kit | Qiagen | 28104 | |
| SCFA filter | Nalgene | 190-2545 | |
| Sony Cell sorter | Sony | ||
| SPRI beads (Agencourt AMPure XP beads) | Beckman | A63881 | |
| Streptavidin-coated microtitre plates | Nalgene | 734-1284 | |
| Streptavidin-PE | Biolegend | 405204 |
Références
- Wright, G. J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems. 5 (12), 1405-1412 (2009).
- van der Merwe, P. A., Barclay, A. N. Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends in Biochemical Sciences. 19 (9), 354-358 (1994).
- Wood, L., Wright, G. J. Approaches to identify extracellular receptor-ligand interactions. Current Opinion in Structural Biology. 56, 28-36 (2019).
- Bushell, K. M., Söllner, C., Schuster-Boeckler, B., Bateman, A., Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Research. 18 (4), 622-630 (2008).
- Visser, J. J., et al. An extracellular biochemical screen reveals that FLRTs and Unc5s mediate neuronal subtype recognition in the retina. eLife. 4, e08149 (2015).
- Özkan, E., et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell. 154 (1), 228-239 (2013).
- Martinez-Martin, N., et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell. 174 (5), 1158-1171 (2018).
- Bianchi, E., Doe, B., Goulding, D., Wright, G. J. Juno is the egg Izumo receptor and is essential for mammalian fertilization. Nature. 508 (7497), 483-487 (2014).
- Mullican, S. E., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature Medicine. 23 (10), 1150-1157 (2017).
- Turner, L., et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 498 (7455), 502-505 (2013).
- Frei, A. P., et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nature Biotechnology. 30 (10), 997-1001 (2012).
- Sobotzki, N., et al. HATRIC-based identification of receptors for orphan ligands. Nature Communications. 9 (1), 1519 (2018).
- Sharma, S., Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 19 (4), (2018).
- Gebre, M., Nomburg, J. L., Gewurz, B. E. CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses. 10 (2), (2018).
- Zotova, A., Zotov, I., Filatov, A., Mazurov, D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. Journal of Immunological Methods. 439, 8-14 (2016).
- Puschnik, A. S., Majzoub, K., Ooi, Y. S., Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nature Reviews. Microbiology. 15 (6), 351-364 (2017).
- Kerr, J. S., Wright, G. J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. Journal of Visualized Experiments. (61), e3881 (2012).
- Tzelepis, K., et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Reports. 17 (4), 1193-1205 (2016).
- Sharma, S., Bartholdson, S. J., Couch, A. C. M., Yusa, K., Wright, G. J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Research. 28 (9), 1372-1382 (2018).
- Wang, B., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nature Protocols. 14 (3), 756-780 (2019).
- Hart, T., et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3. 7 (8), 2719-2727 (2017).
- Kuleshov, M. V., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44 (W1), W90-W97 (2016).
- Crosnier, C., et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 480 (7378), 534-537 (2011).
- Kirk, P., et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal. 19 (15), 3896-3904 (2000).
- Chong, Z. S., Ohnishi, S., Yusa, K., Wright, G. J. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biology. 19 (1), 205 (2018).
- van der Meer, D., et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Research. 47 (D1), D923-D929 (2019).
- Bausch-Fluck, D., et al. A mass spectrometric-derived cell surface protein atlas. PloS One. 10 (3), e0121314 (2015).
- Behan, F. M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 568 (7753), 511-516 (2019).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.