
Method Article
Cell Surface Receptor Identification Using Genome-Scale CRISPR/Cas9 Genetic Screens
In This Article
Summary
This manuscript describes a genome-scale cell-based screening approach to identify extracellular receptor-ligand interactions.
Abstract
Intercellular communication mediated by direct interactions between membrane-embedded cell surface receptors is crucial for the normal development and functioning of multicellular organisms. Detecting these interactions remains technically challenging, however. This manuscript describes a systematic genome-scale CRISPR/Cas9 knockout genetic screening approach that reveals cellular pathways required for specific cell surface recognition events. This assay utilizes recombinant proteins produced in a mammalian protein expression system as avid binding probes to identify interaction partners in a cell-based genetic screen. This method can be used to identify the genes necessary for cell surface interactions detected by recombinant binding probes corresponding to the ectodomains of membrane-embedded receptors. Importantly, given the genome-scale nature of this approach, it also has the advantage of not only identifying the direct receptor but also the cellular components that are required for the presentation of the receptor at the cell surface, thereby providing valuable insights into the biology of the receptor.
Introduction
Extracellular interactions by cell surface receptor proteins direct important biological processes such as tissue organization, host-pathogen recognition, and immune regulation. Investigating these interactions is of interest to the wider biomedical community, because membrane receptors are actionable targets of systematically delivered therapeutics such as monoclonal antibodies. Despite their importance, studying these interactions remains technically challenging. This is mainly because membrane-embedded receptors are amphipathic, making them difficult to isolate from biological membranes for biochemical manipulation, and their interactions are typified by the weak interaction affinities (KDs in the µM-mM range)1. Consequently, many commonly used methods are unsuitable to detect this class of protein interactions1,2.
A range of methods has been developed to specifically investigate extracellular receptor-ligand interactions that take their unique biochemical properties into consideration3. A number of these approaches involve expressing the entire ectodomain of a receptor as a soluble recombinant protein in mammalian or insect cell-based systems to ensure that these proteins contain posttranslational modifications that are structurally important, such as glycans and disulfide bonds. To overcome the low-affinity binding, the ectodomains are often oligomerized to increase their binding avidity. Avid protein ectodomains have been successfully used as binding probes to identify interaction partners in direct recombinant protein-protein interaction screens4,5,6,7. While broadly successful, recombinant protein-based methods require that the ectodomain of a membrane receptor be produced as a soluble protein. Therefore, it is only generally applicable to proteins that contain a contiguous extracellular region (e.g., single-pass type I, type II, or GPI-anchored) and is not generally suitable for receptor complexes and membrane proteins that span the membrane multiple times.
Expression cloning techniques in which a library of complementary DNAs (cDNAs) is transfected into cells and tested for a gain-of-binding phenotype have also been used to identify extracellular protein-protein interactions8. The availability of large collections of cloned and sequenced cDNA expression plasmids in recent years has facilitated methods in which cell lines overexpressing cDNAs encoding cell surface receptors are screened for the binding of recombinant proteins to identify interactions9,10. The cDNA overexpression-based approaches, unlike recombinant protein-based methods, afford the possibility to identify interactions in the context of the plasma membrane. However, the success of using cDNA expression constructs depends on the cells' ability to overexpress the protein in the correctly folded form, but this often requires cellular accessory factors such as transporters, chaperones, and correct oligomeric assembly. Transfecting a single cDNA might therefore not be enough to achieve cell surface expression.
Screening techniques using cDNA constructs or recombinant protein probes are resource-intensive and require large collections of cDNA or recombinant protein libraries. Specifically designed mass spectrometry-based methods have been utilized recently to identify extracellular interactions that do not require the assembly of large libraries. However, these techniques require chemical manipulation of the cell surface, which can alter the biochemical nature of the molecules present on the surface of the cells and are currently only applicable for interactions mediated by glycosylated proteins11,12. The majority of the currently available methods also heavily focus on the interactions between proteins while largely ignoring the contribution from the membrane microenvironment, including molecules such as glycans, lipids, and cholesterol.
The recent development of highly efficient bialleleic targeting using CRISPR-based approaches has enabled genome-scale libraries of cells lacking defined genes in a single pool that can be screened in a systematic and unbiased way to identify cellular components involved in different contexts, including dissecting cellular signaling processes, identification of perturbations that confer resistance to drugs, toxins, and pathogens, and determining specificity of antibodies13,14,15,16. Here, we describe a genome-scale CRISPR-based knockout cell screening assay that provides an alternative to the current biochemical approaches to identify extracellular receptor-ligand interactions. This approach of identifying interactions mediated by membrane receptors by genetic screens is particularly suitable for researchers that have a focused interest on individual ligands because it avoids the need to compile large libraries of cDNAs or recombinant proteins.
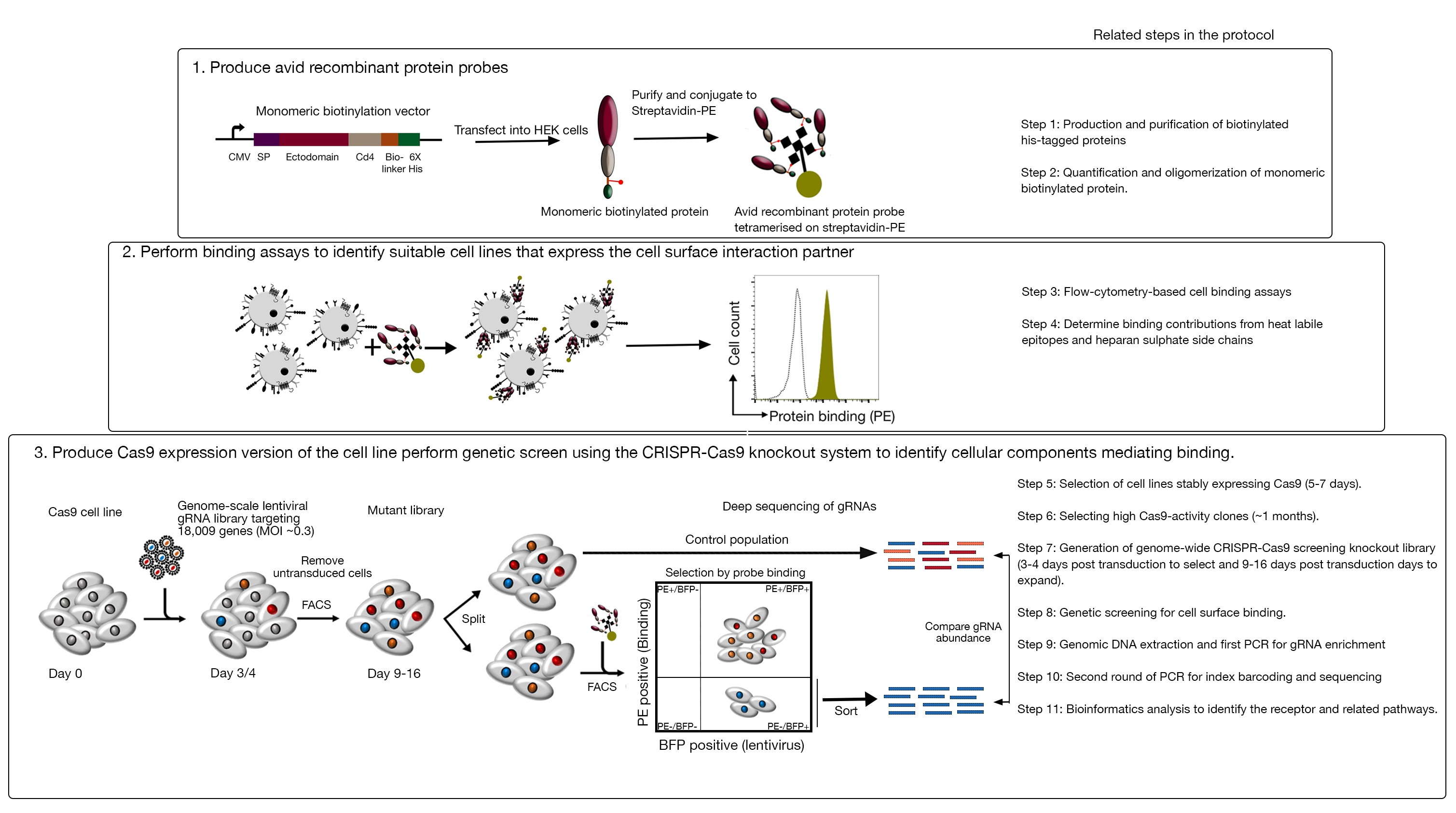
This assay consists of three major steps: 1) Highly avid recombinant protein binding probes consisting of the extracellular regions of a receptor of interest are produced and used in fluorescence-based flow cytometry-based binding assays; 2) The binding assays are used to identify a cell line that expresses the interaction partner of the recombinant protein probe; 3) A Cas9-expressing version of the cell line that interacts with the protein of interest is produced and a genome-scale CRISPR/Cas9-based knockout screen is performed (Figure 1). In this genetic screen, binding of a recombinant protein to cell lines is used as a measurable phenotype in which cells within the knockout library that have lost the ability to bind the probe are sorted using fluorescence-based activated cell sorting (FACS) and the genes that caused the loss of the binding phenotype identified by sequencing. In principle, the genes encoding the receptor responsible for binding the avid probe and those required for its cell surface display are identified.
The first step of this protocol involves the production of avid recombinant protein probes representing the ectodomain of the membrane-bound receptors. These receptors are known to frequently retain their extracellular binding functions when their ectodomains are expressed as a recombinant soluble protein1. For a protein of interest, soluble recombinant proteins can be produced in any suitable eukaryotic protein expression system in any format provided that it can be oligomerized for increased binding avidity, and it contains tags that can be used in fluorescence-based flow cytometry-based binding assays (e.g., FLAG-tag, biotin-tag). Detailed protocols for the production of soluble ectodomains of membrane receptors using the HEK293 protein expression system, as well as different multimerization techniques and the protein expression constructs for the production of both pentameric proteins and monomeric proteins have been previously described1,17. The protocol here will describe the steps for generating fluorescent avid probes from monomeric biotinylated proteins by conjugating them to streptavidin conjugated to a fluorochrome (e.g., phycoerythrin, or PE), which can be used directly in cell-based binding assays and has the advantage of not requiring a secondary antibody for detection. General protocols for performing genome-scale screens have already been described20,21, thus the protocol mainly focus on the specifics of performing flow cytometry-based recombinant protein binding screens using the CRISPR/Cas9 knockout screening system using the Human V1 ("Yusa") library18.
Protocol
1. Production and purification of biotinylated His-tagged proteins
- Use a mammalian or insect cell-based protein expression system to produce soluble recombinant His-tagged biotinylated proteins (see plasmid constructs in Table 1).
NOTE: A detailed protocol for the production of monomeric biotin and His-tagged proteins using the HEK293 cell expression system is described by Kerr et al.17. Protein ectodomains expressed using the HEK293 expression system are secreted into the culture medium. - Collect the soluble proteins by pelleting the cells by centrifugation at 3,000 x g for 20 min.
- Filter the supernatant through a 0.22 µm filter and add the Ni2+-NTA agarose beads to the filtered protein supernatant in a 1:1,000 ratio (i.e., 50 µL of 50% agarose slurry into 50 mL of supernatant). Incubate overnight or at least 4-5 h at 4 °C on a rotating platform.
- Wash the polypropylene column by adding 5 mL of His-purification wash buffer. Refer to Table 2 for all buffer compositions.
- Pour the entire bead-protein supernatant mixture into the column. Beads will accumulate at the base.
- Wash the beads 2x with 15 mL of wash buffer. To avoid protein dilution, carefully draw the residual wash buffer from the column with a 5 mL syringe and discard.
- Carefully add 300-500 µL of His-purification elution buffer directly to the beads and incubate for at least 1 h. Collect the eluted protein by again carefully drawing out the liquid using a 1 mL syringe. Exchange the elution buffer to the desired buffer (e.g., normally PBS or HBS) using desalting columns. Store all proteins at 4 °C until further use.
2. Quantification and oligomerization of monomeric biotinylated protein
NOTE: To increase the binding avidity, oligomerize biotinylated monomeric proteins on tetrameric streptavidin-PE before using them in binding assays. Achieve optimal conjugation ratios of monomeric proteins and tetrameric streptavidin-PE by testing a dilution series of biotinylated monomers against a fixed concentration of streptavidin and by empirically establishing the minimum dilution at which no excess biotinylated monomers can be detected.
- Make at least eight serial dilutions of biotinylated protein samples using an appropriate dilution buffer (either PBS or HBS with 1% bovine serum albumin [BSA]) in a 96 well plate. Ensure that the final volume of each dilution is at least 200 µL.
- Make a duplicate plate of the samples by removing 100 µL from each well and transferring into a new 96 well plate. Always include a control. In this case controls are tag-only proteins (i.e., biotinylated His-tagged Cd4 domain 3+4 protein). This will be used as a control probe in all binding assays.
- Dilute streptavidin-PE to 0.1 µg/mL in the dilution buffer.
- On just one of the plates, add 100 µL of the diluted streptavidin-PE. The duplicate plate will serve as a control. Add 100 µL of dilution buffer in the control plate to equalize volumes.
- Incubate for 20 min at room temperature (RT). In the meantime, block the wells of a streptavidin-coated plate with the dilution buffer for 15 min.
- Transfer the total volume of the sample from both plates to individual wells of the streptavidin-coated plates and incubate for 1 h at RT.
- Wash the plate 3x with 200 µL of wash buffer (i.e., either PBS or HBS with 0.1% Tween-20, 2% BSA). Add 100 µL of 2 µg/mL mouse anti-rat Cd4d3+4 IgG (OX68) and incubate for 1 h at RT.
- Wash the plate 3x with the wash buffer. Add 100 µL of an anti-mouse alkaline phosphatase conjugate at 0.2 µg/mL for 1 h at RT.
- Wash the plate 3x with wash buffer and 1x in dilution buffer.
- Prepare p-nitrophenyl phosphate at 1 mg/mL in diethanolamine buffer. Add 100 µL in each well and incubate for 15 min.
- Take absorbance readings at 405 nm. Use the minimum dilution at which there is no signal on the plate as the appropriate dilution factor to create tetramers (Figure 2).
- Make a 10x tetramer staining solution for all samples and controls by incubating 4 µg/mL streptavidin-PE and the appropriate biotinylated protein dilution for 30 min at RT. Store conjugated proteins in a light-protected tube at 4 °C until further use.
3. Flow cytometry-based cell binding assays
- For adherent cells, remove culture media and wash 1x with PBS without divalent cations. Then add cell detachment solutions (e.g., EDTA). Allow the cells to detach for 5-10 min. Gently tap the flask to release the cells.
NOTE: Avoid using trypsin-based products as they can cleave cell surface proteins. - Collect detached cells into a tube. For cells growing in suspension (e.g., HEK293 cells), directly collect the cells from culture flasks into a tube.
- Pellet cells at 200 x g for 5 min. Remove the supernatant and resuspend the pellet in wash buffer (i.e., PBS/1% BSA).
- Count the cells using a hemocytometer and adjust the concentration to 2.5 x 105-1 x 106 cells/mL. Aliquot 100 µL of prepared cell mix on a 96 well U- or V- bottomed plate. Spin the plate for 5 min at 400 x g. Remove the supernatant with a multichannel pipette.
- Add 100 µL of normalized fluorescently labeled highly avid protein probes and controls into the previously prepared plates with cells and incubate for 1 h at 4 °C. After binding for 1 h, spin the plate at 400 x g for 5 min.
- Remove the supernatant and add 200 µL of wash buffer (i.e., PBS/1% BSA). Mix well by pipetting up and down.
- Pellet the cells by centrifugation at 400 x g for 5 min. Repeat the wash step 1x. After two washes, completely remove the supernatant and resuspend the cell pellet in 100 µL of PBS.
- Analyze the cells by flow cytometry. Use the yellow-green laser (i.e., 561 nm) to detect PE fluorescence.
- First analyze the cells that have been stained with the control probe. Based on the distribution of PE fluorescence, draw a gate for binding population such that no more than 1% of the control cell falls in this gate.
- Analyze the sample and determine the fraction of cells that falls in the binding gate.
NOTE: Cell lines that display a higher binding population are desired for genetic screens, as they have a higher signal-to-noise ratio. Ideally over 80% of the cells should fall within this gate.
4. Determining binding contributions from heat labile epitopes and heparan sulphate sidechains
NOTE: The activity of many proteins is heat labile, so loss of binding activity following heat treatment is encouraging. It is advised to determine the contribution from negatively charged glycosaminoglycans, mainly heparan sulphate (HS), in mediating binding of the recombinant proteins. This is because the binding by HS in the cell binding assay described here can be additive rather than codependent on other receptors19. This means that the observed binding could be entirely mediated by HS side chains of cell surface proteoglycans and not by a specific receptor. Binding to HS on the cell surface is not necessarily nonspecific, but rather a property of a protein, which is useful to know before performing a full genetic screen.
- Prepare heat-treated protein samples to use in binding assays.
- Heat the normalized but unconjugated monomeric protein at 80 °C for 10 min.
- Conjugate the heat-treated protein to streptavidin-PE assuming the same conjugation ratio as its untreated counterpart as determined by ELISA (refer to section 2).
- Prepare heparin-blocked protein samples.
- Prepare eight 1:3 dilutions of soluble heparin in PBS with a starting concentration of 2 mg/mL and final volume of 100 µL.
- Incubate 100 µL of prepared binding probes in the heparin dilutions for at least 30 min.
- Use heat-treated protein and the full 200 µL of heparin/protein mixture in the binding assays described in section 3. Representative results are shown in Figure 3A,B.
5. Selection of cell lines stably expressing Cas9
NOTE: Before the cell line that binds the probe of interest can be used in CRISPR screening, it must first be engineered to express the Cas9 nuclease and a highly active clone selected19.
- Use the following general lentivirus production protocol to produce lentivirus using the lentiviral construct for Cas9 expression (refer to Table 1).
- Culture HEK293-FT cells in DMEM/10% FBS media at 37 °C and 5% CO2. Seed HEK293-FT cells 1 day prior to transfection so that they are ~80% confluent on the day of transfection.
NOTE: HEK293FT cells are loosely adherent; therefore, when they are used for production of lentiviruses, consider plating them on culture flasks coated with 0.1% (w/v) gelatin to increase adherence. - Perform transfections in the morning. Add transfer vector, packaging mix, and transfection reagent into prewarmed transfection compatible media (e.g., Opti-MEM). Mix by inverting the tube 10-15x. Incubate for 5 min at RT. Refer to Table 3 for exact volumes.
- Add the transfection reagent as suggested by the manufacturer. Mix by quick vortexing. Incubate for 30 min at RT.
- Very carefully aspirate the spent medium. Add transfection compatible media to plate.
- Add the transfection reagent/DNA complexes dropwise onto the side of the plate and slowly spread through the plate by swirling very gently.
- Incubate at 37 °C for 3-5 h and replace the medium with D10 medium. Incubate overnight.
- Next day in the morning, replace the medium with fresh D10 medium. Incubate overnight.
- Next day late in the afternoon, collect the viral supernatant. Filter with a 0.45 µm filter with low protein binding. Optionally, add fresh D10 medium, incubate overnight and recollect the supernatant the next day.
- Virus supernatants are stable at 4 °C for only for a few days. Store at -80 °C for long-term storage.
NOTE: To generate a highly concentrated lentiviral preparation, which could be desirable for the transduction of difficult to transduce cells, supernatants can also be concentrated by centrifugation at 6,000 x g overnight at 4 °C. Mark the translucent viral pellet with an ethanol-resistant pen and discard the supernatant. Resuspend the pellet in 1/100th of the original volume for a 100x increase in the concentration.
- Culture HEK293-FT cells in DMEM/10% FBS media at 37 °C and 5% CO2. Seed HEK293-FT cells 1 day prior to transfection so that they are ~80% confluent on the day of transfection.
- Transduce the cells with lentiviruses.
- Plate 1 x 106 cells per well in a 6 well plate with 3 mL of appropriate culture media. Some cells are more readily transduced than others. For easy to transduce cells (e.g., HEK cells), directly add the lentivirus to the cells. For difficult to transduce cells, it might be necessary to follow a spinoculation protocol as described below.
- Aliquot 2 mL of 2-5 x 106 cells/mL in a 15 mL conical tube.
- Add lentivirus together with 8 µg/mL hexadimethrine bromide and incubate at RT for 30 min.
- Centrifuge for 100 min at 800 x g at 32 °C. Then resuspended the cells in the same media and add the cell suspension into appropriate culture flasks with appropriate media.
- Allow transductions for at least 24 h. Afterwards remove the media containing the virus and add fresh medium.
- After another 24 h, change the media to one that is supplemented with the appropriate antibiotics. The Cas9 construct contains a blasticidin resistance cassette for selection.
NOTE: The amount of blasticidin must be optimized for each cell line by performing a dose response kill curve. A blasticidin concentration between 2.5-50 µg/mL should kill most untransduced cell lines within 10 days of transduction.
- Plate 1 x 106 cells per well in a 6 well plate with 3 mL of appropriate culture media. Some cells are more readily transduced than others. For easy to transduce cells (e.g., HEK cells), directly add the lentivirus to the cells. For difficult to transduce cells, it might be necessary to follow a spinoculation protocol as described below.
- Perform selection until all cells in the control plate (i.e., nontransduced cells that have been treated with the same concentration of selection antibiotics) are killed.
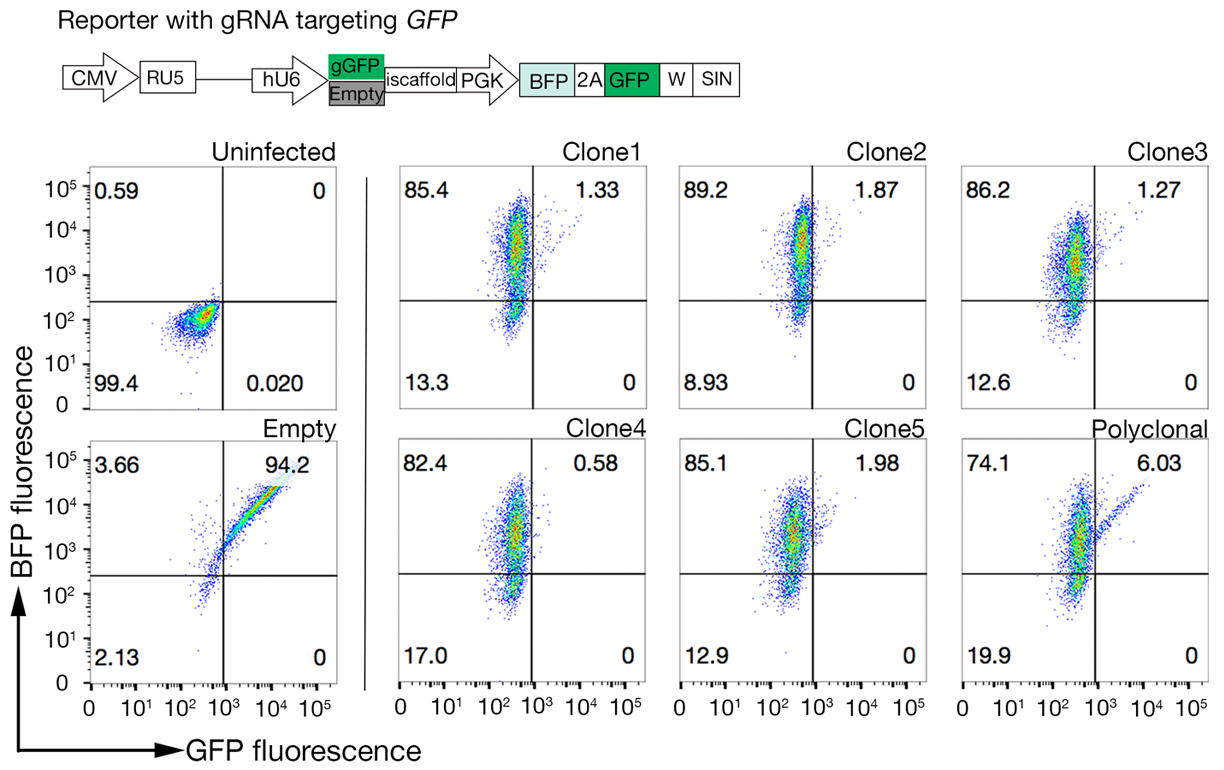
6. Selecting high Cas9-activity clones
NOTE: Polyclonal Cas9 can be used to successfully perform genetic screens; however, selecting a clone with high Cas9 activity improves the screening results18.
- Use limiting dilution or single-cell sort individual blasticidin-resistant cells into wells of three 96 well plates containing culture media supplemented with blasticidin. Clones will start to emerge between 2-4 weeks. Select 10-20 clones and expand in 6 well plates.
- Assay the clones for Cas9 activity using the quick-to-assess GFP-BFP (green fluorescent protein-blue fluorescent protein) system, which uses an exogenous gene knockout system in which cells are transduced either with a construct expressing GFP with a gRNA-targeting GFP or an empty gRNA as a control18.
- Order reporter plasmids: GFP-BFP plasmid, Control-BFP plasmid (Table 1).
- Produce lentivirus for both the GFP-BFP plasmid and Control-BFP plasmid using the lentivirus production protocol described in section 5.1.
- Transduce each Cas9-expressing cell line clone with the lentivirus encoding the GFP-BFP system and Control-BFP separately. Follow the protocol in section 5.2.
- After 3 days of transduction, examine the GFP-BFP fluorescence of each clone using flow cytometry. Use 488 nm laser and 405 nm laser to detect GFP and BFP respectively.
- Quantitate the Cas9 activity in each clone by examining the ratio of BFP only to GFP-BFP-double positive cells. High activity Cas9 cells should ideally have >95% GFP knockout efficiency (Figure 4).
7. Generation of genome-wide CRISPR-Cas9 screening knockout library
- For the genome-wide screening using the Human V1 library18, order the genome-wide library (refer to Table 1) and prepare the plasmid library from the bacterial stab using the protocol provided under "Protocols for Library Replication" in the manufacturer's manual.
- Use the genome-wide library plasmid preparation to produce a lentiviral library encoding gRNAs for targeted disruption of human genes using the lentivirus production protocol described in section 5.1.
NOTE: A good practice is to produce a single batch of lentiviral preparation that is optimized for transduction to improve experimental consistency. - Use the transduction protocol in section 5.2 to perform small scale test transductions to determine the required amount of virus for each cell line to achieve 30% transduction. Use flow cytometry to assess BFP fluorescence as a proxy for transduction efficiency.
- To transduce HEK293 cells, simply add the predetermined lentiviral preparation to 30-50 x 106 cells cultured in normal growth media for ~4 h. Then remove the media with lentivirus and replace with fresh growth media.
- For other cell lines, use the spinoculation protocol in section 5.2.1 but at a larger scale, such that a total of 30-50 x 106 cells are transduced. For this, aliquot 2 mL of 5 x 106 cells/mL in a 15 mL conical tube and proceed as indicated.
- For adherent cell lines, select transduced cells by adding puromycin 24 h after transduction.
NOTE: Optimize puromycin concentrations by performing a dose response kill curve. Normally concentrations between 1-10 µg/mL should kill nontransduced cells within 3-5 days. Avoid using higher concentrations of puromycin because this may increase the chances of selecting cells that have been transduced by more than onesingle guide RNA (sgRNA). - For suspension cells, harvest transduced (i.e., BFP positive) cells 3 days posttransduction using a cell sorter and generate libraries that contain at least 10 x 106 cells. Once selected using BFP, grow the cells in media supplemented with appropriate amount of puromycin.
NOTE: Avoid selections only with puromycin for suspension cell lines, because it is difficult to remove dead cells and debris from suspension cell cultures that can interfere with cell sorting. - Culture mutant library for 9-16 days posttransduction with regular passage every 2-3 days.
8. Genetic screening for cell surface binding
- Pellet the mutant cell library at 200 x g for 5 min and resuspend the cells in PBS.
- Divide the cells into two 15 mL conical tubes with at least 50 x 106 cells in each tube.
- Spin one conical tube at 200 x g for 5 min, remove the supernatant, and freeze the cell pellet at -20 °C. This is the control population and will be processed later.
- Resuspend the pellet in the other tube in 10 mL of PBS/1% BSA. Set aside 100 µL of cells as a negative control on a 96 well plate.
- Add the appropriate preconjugated recombinant protein to the cell suspension in the conical tube and negative control proteins to the 96 well plate.
- Perform cell staining for at least 1 h at 4 °C on a benchtop rotor with gentle rotation (6 rpm).
- Pellet the cells at 200 x g for 5 min, remove the supernatant. Perform two wash steps, then resuspend the cells in 5 mL of PBS.
- Strain the cells though a 30 µm cell strainer to remove cell clusters. Analyze using a flow sorter.
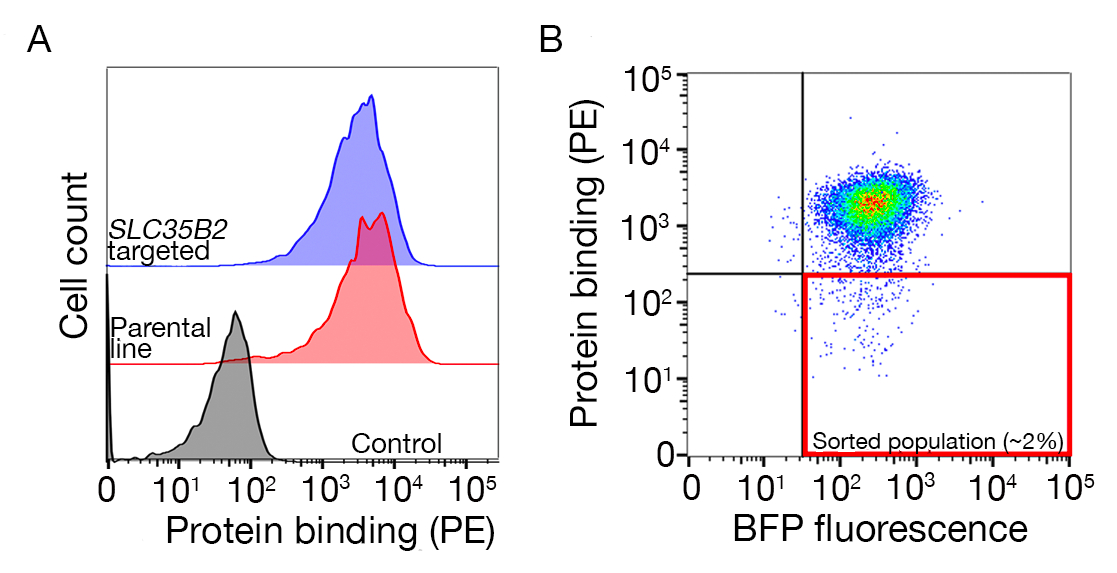
- Use the negative control sample to gate for BFP+/PE- cells.
- Sort the sample and collect the BFP+/PE- cells. The sort gates will depend on the binding of the cells to the protein but is normally 1-5% of the PE negative samples are collected. An example of a sorting gate is provided in Supplementary Figure 1.
- Collect 500,000-1,000,000 cells from the selected gate. Given the low number of cells, consider collecting the samples in a 1.5 mL centrifugation tube to minimize losses.
- Pellet the sorted cells by centrifuging at 500 x g for 5 min. Carefully remove the supernatant and discard. It is possible to store the pellet at -20 °C for up to 6 months.
9. Genomic DNA extraction and first PCR for gRNA enrichment
- Extract genomic DNA from high-complexity control population.
- If the control population was frozen at -20 °C, take out the conical tube and add PBS. Keep on ice to thaw the pellet.
- Use a commercial kit (see Table of Materials) using the manufacturer's recommendations to extract genomic DNA from 50 x 106 cells. Adjust the DNA concentration to 1 mg/mL.
- For each sample, set up a master mix for PCR corresponding to 72 µg of DNA. Aliquot 50 µL per well in 36 wells of a 96 well PCR plate. The necessary primer sequences are listed in Table 4. Use the guide in Table 5 and 6.
- Resolve 5 µL of the PCR from 6-12 representative samples on a 2% (w/v) agarose gel. A single clear band at ~250 bp should be observed. If the bands are faint, repeat the PCR for an additional 2-3 cycles.
- Use a multichannel pipette to collect 5 µL of PCR products from each well (180 µL in total) and pool them in a reservoir with 900 µL of binding buffer from a commercial kit (see Table of Materials).
- Purify the PCR products using a commercial PCR purification kit. Elute DNA into 50 µL of elution buffer from a commercial kit (see Table of Materials) and measure the DNA concentration.
- Samples that have been sorted for the loss of binding phenotype are unlikely to be composed of a large number of independent clones. Therefore, it is not necessary to perform PCR with 72 µg of DNA. Isolate the DNA using an appropriate commercial kit (see Table of Materials). Set up 3-4 PCR reactions using the protocol described before (section 9.1.3) with 100 ng/µL DNA. If the sorted cell number is less than 100,000 use cell lysates instead of genomic DNA preparations.
- Aliquot approximately 10,000 cells/well in a 96 well PCR plate.
- Pellet the cells in the plate and carefully remove most of the supernatant. The pellet will not be visible.
- Add 25 µL of water in each well and heat the samples at 95 °C for 10 min.
- Add 5 µL of 2 mg/mL freshly diluted proteinase K to each well for 1 h and incubate at 56 °C. Then heat the sample for 10 min at 95 °C to inactivate the proteinase K.
- Use 10 µL of cell lysate mixture per PCR reaction. Lysates should be used within 24 h.
10. Second round of PCR for index barcoding and sequencing
- Dilute the products from the first round PCR to 40 pg/µL.
- Set up one PCR per sample (use the guide provided in Tables 7 and 8). The use of a high-fidelity polymerase is important to minimize errors introduced by the polymerase during sgRNA amplification.
- Resolve 5 µL of PCR products on a 2% (w/v) agarose gel. A single clear band at ~330 bps should be observed.
- Purify PCR products using paramagnetic beads by adding 31.5 µL of (0.7x total volume) of resuspended beads to the PCR products, mixing well, and incubating for 5 min at RT.
- Place the tube on a magnetic rack for 3 min. The beads should be captured on the side of the plate and the solution should be clear. Carefully remove and discard the supernatant.
- Add 150 µl of 80% freshly prepared ethanol to the tube. Incubate for 30 s, and then carefully remove and discard the supernatant.
- Repeat step 13.6, this time with 180 µL. Then air dry the beads for 5 min.
- Remove the tube from the magnet. Elute DNA target from beads into 35 µL of sterile EB buffer. Incubate for 3 min, then put the tube back in the magnetic rack for 3 min.
- Transfer approximately 30 µL of the supernatant containing the eluted PCR products to a clean tube.
- Sequence the samples on a next-generation sequencing platform. For the HumanV1 gRNA library, use the custom primer listed in Table 4 to sequence 19 bp.
11. Bioinformatics analysis to identify the receptor and related pathways
- Map sequences from sorted and unsorted population to the reference library using the count function of MAGeCK. The function will yield a raw count file (Supplementary Table 1).
NOTE: Detailed instructions on the installation of MAGeCK and the usage of different functions within MAGeCK are described in a previously published protocol by Wang et al.20. - Check the technical standard of the control library used in the screen.
- Median normalize the raw counts and use the ggplot2 package in R21 or equivalent software to plot an empirical cumulative density function plot of the counts in plasmid and control unsorted samples (Figure 5A).
- Run the -test function of MAGeCK using counts from plasmid population as "control" and the counts from unsorted control samples as the "test" sample. The function is will yield a gene summary file (Supplementary Table 1).
- Open the gene summary file and draw the distribution of log-fold-changes (neg|lfc column) for previously categorized essential and nonessential genes22 (Figure 5B).
- Select significantly depleted genes (neg|fdr < 0.05) and perform pathway enrichment analysis using the enrichr23 package or any equivalent pathway enrichment packages in R (Figure 5C).
- Run the -test function of MAGeCK with default setting. Use raw counts from unsorted control sample as "control" and counts from sorted sample as "treatment" when performing the analysis.
- Open the gene summary file generated by MAGeCK and rank the pos|rank column in ascending order. Use FDR (pos|fdr column) < 0.05 as a cutoff for identification of hits. The receptor is usually ranked highly, often in the first position.
- Plot the Robust-Ranking-Algorithm (RRA) scores for positive selection (pos|score) in R or an equivalent software (Figure 5D).
- Select the gene hits and perform pathway enrichment analysis using the enrichr package or any equivalent pathway enrichment packages in R to identify the enriched pathways.
Results
Sequencing data from two representative genome-scale knockout screens for the identification of the binding partner of human TNFSF9 and P. falciparum RH5 performed in NCI-SNU-1 and HEK293 cells respectively are provided (Supplementary Table 1). The binding behavior of RH5 was affected by both heparan sulphate and its known receptor BSG24 (Figure 3C), whereas TNFRSF9 specifically bound to its known receptor TNFSF9 and did not lose binding upon preincubation with soluble heparin. Protein 3 in Figure 3B represents TNFRSF9.
For both cell lines, the distribution of gRNAs in the control mutant library after 3 days (9, 14, and 16 days posttransduction) are also provided (Supplementary Table 1). The gRNA distribution revealed that the library complexity was maintained throughout the course of the experiment (Figure 5A). The genetic screen for the identification of the ligand for TNFSF9 was performed on day 14 posttransduction, whereas that for RH5 was performed day 9 posttransduction. The technical quality of the screens was assessed by examining the distribution of observed fold-changes of gRNAs targeting a reference set of nonessential genes compared to the distribution for reference set of essential genes22 (Figure 5B). In addition, pathway-level enrichment also revealed that expected essential pathways were identified and significantly enriched in the "drop-out" population when comparing the control sample to the original plasmid library. An example with day 14 NCI-SNU-1 sample is depicted in Figure 5C.
The distribution of the gRNAs in the control versus sorted population using the -test function of MAGeCK (see Supplementary Table 1 for the gene summary output from MAGeCK) was used to identify the receptor from the phenotypic screens. The modified RRA score reported by MAGeCK in the gene-level analysis is plotted against the genes ranked by p values. The RRA score in MAGeCK provides a measure in which gRNAs are ranked consistently higher than expected. In the screen for TNFRSF9, the top hit was TNFSF9, which is a known binding partner of TNFRSF9 (Figure 5D). In addition, a number of genes related to the TP53 pathway were also identified. In the case of RH5, in addition to the known receptor (BSG) and the gene required for the production of the sulfated GAGs (SLC35B2), an additional gene (SLC16A1) was also identified (Figure 5E). SLC16A1 is a chaperone required for trafficking BSG to the surface of cells25. Together, these results demonstrate the ability of the screen to identify directly interacting receptors and the cellular components required for that receptor to be expressed on the surface of the cells in a functional form.

Figure 1: Overview of the genetic screening approach to identify cell surface receptors. This assay consists of three major steps: First, recombinant proteins representing the ectodomain of cell surface receptors are expressed in a cell line that can add structurally critical posttranslational modifications such as HEK293 cells. Monomeric protein ectodomains are oligomerized by conjugating to streptavidin-PE to increase their binding avidity. Second, these avid probes are used in cellular binding assays where bright staining on the cell lines indicated by a prominent shift in PE fluorescence (in green) compared to a negative control protein (in black) demonstrates the presence of a cell surface binding partner. Third, receptor-positive Cas9-expressing cell lines are selected and genome-scale screening using gRNAs targeting the vast majority of protein-coding genes is performed. While generating mutant libraries, it is common to use 30% transduction efficiency, which is based on the Poisson distribution probability that ensures each cell receives a single gRNA such that the resultant phenotype is attributed to a specific knockout. The BFP marker expressed by the transduced cells is used to select cells containing gRNAs using FACS. Phenotypic screens are performed between 9-16 days posttransduction. On the day of the screen, the total mutant cell population is divided into two. One half is kept as the control population and the other half is selected for recombinant protein binding. The cells from the mutant library that are no longer able to bind the recombinant protein are sorted using FACS and the enrichment of gRNAs in the sorted versus control population is used to identify genes required for cell surface binding of the labeled avid probe. Steps in the protocol that require considerable time are indicated. This figure has been modified from Sharma et al.19. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Establishing the ratios of biotinylated protein to streptavidin-PE using an ELISA-based method. An example of streptavidin-PE conjugation strategy used to generate an avid probe from a biotinylated monomeric protein. A dilution series of biotinylated monomers was incubated against a fixed concentration of streptavidin. The minimum dilution at which no excess biotinylated monomers can be detected was determined by ELISA. ELISA was performed with or without preincubating a range of protein dilutions with 10 ng of streptavidin-PE. In the presence of streptavidin-PE, the minimum dilution at which no signal was identified (circled black) and the amount of protein required for the saturation was calculated to generate a 10x stock solution with 4 µg/mL streptavidin-PE. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative binding of proteins to cell lines. (A) Protein binding to cell lines had a clear increase in cell-associated fluorescence compared to the control sample. Heat treatment (80 °C for 10 min) of recombinant protein abrogated all binding back to a negative control, demonstrating that the binding behavior was dependent on correctly folded protein. (B) Different classes of protein binding behavior to cell surfaces; dependence on GAGs. From left to right, the proteins can be classified into three types: Protein type 1 only adsorbs to HS. These proteins lose their binding after preincubation with heparin concentrations over 0.2 mg/mL. Protein type 2 binds to HS in addition to a specific receptor. These proteins lose partial binding in the preblocking experiments. Protein type 3 does not bind HS. These proteins do not lose binding compared to parental lines. (C) An example of a protein (i.e., RH5) that binds to HS and a specific receptor in an additive manner. Targeting either the receptor (i.e., BSG) or enzymes required for HS synthesis (e.g., SLC35B2, EXTL3) only partially reduces the binding of RH5 to cells relative to controls. Transduced polyclonal lines can be used in such experiments to establish binding behavior. This figure has been modified from Sharma et al.19. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Selecting clonal cell lines with high Cas9 activity. Genome-editing efficiency of both polyclonal and cloned lines of NCI-SNU-1 cell lines were assessed using the GFP-BFP reporter system, in which cell lines were transduced with viruses with a gRNA-targeting plasmid encoded GFP or without (i.e., "empty"). A schematic is depicted. Flow cytometry was used to test both BFP and GFP expression after transduction and compared to uninfected control. GFP expression was used as a proxy for Cas9 activity, whereas BFP expression marked transduced cells. The profile for uninfected and empty infected cells looked similar for all clones. Representative profiles are depicted in the left panel. All five clones of the NCI-SNU-1 cell line showed a higher loss of GFP compared to the polyclonal line (right panel), with clone 4 showing the highest efficiency with the lowest refractory population. This figure has been modified from Sharma et al.19. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative results from genetic screens for the identification of the cell surface binding partners. (A) Cumulative distribution function plots comparing the gRNA abundance in the plasmid library to the mutant libraries of HEK-293-E and NCI-SNU-1 cells on day 9, 14, and 16 days posttransduction. For any given number, cumulative density function reports the percent of datapoints that were below that threshold. The small shift of the mutant cell population compared to the original plasmid population represents the depletion in a subset of gRNAs compared to the plasmid library. (B) Distribution of log-fold changes in genes that have been previously categorized as being essential (red) or nonessential (black) in the HEK293 and NCI-SNU-1 cell lines. The distribution of fold-changes for nonessential genes centered at ~0, whereas that for essential genes shifted to the left towards negative fold changes. (C) Significantly enriched pathways in genes depleted in NCI-SNU-1 mutant control population 14 days posttransduction. Expected known cell-essential pathways were identified. (D) Robust Rank Algorithm (RRA)-score for genes that were enriched in the sorted cells that had lost the ability to bind the TNFRSF9 probe. Genes were ranked according to the RRA-score. The known interaction partner TNFSF9 and genes related to the TP53 pathway (labeled in red) were identified in the screen. (E) Rank-ordered RRA-scores for genes identified from gRNA enrichment analysis required for RH5 binding to HEK293 cells (left panel). SLC35B2 and SLC16A1 were identified within a false-discovery-rate (FDR) threshold of 5%. Two additional genes in the HS biosynthesis pathway (i.e., EXTL3 and NDST1) were identified within FDR of 25%. Schematic depicting the general GAG biosynthesis pathway with the relevant genes mapped to the corresponding steps (panel 2). Genes required for the commitment to chondroitin sulphate biogenesis (i.e., CSGALNACT1/2) were not identified in the screen. This figure has been modified from Sharma et al.19. Please click here to view a larger version of this figure.
{kind=link}
| Plasmid name | Plasmid # | Use |
| Protein expression construct: CD200RCD4d3+4-bio-linker-his | Addgene: 36153 | Production of recombinant Protein with CD4d3+4, biotin and 6-his tags. |
| pMD2.G | Addgene: 12259 | VSV-G envelope expressing plasmid; production of lentivirus |
| psPAX2 | Addgene: 12260 | Lentiviral packaging plasmid, production of lentivirus |
| Cas9-construct: pKLV2-EF1a-Cas9Bsd-W | Addgene: 68343 | Production of constitutively expressing Cas9 line |
| gRNA expression construct: pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W | Addgene: 67974 | CRISPR gRNA expression vector with an improved scaffold and puro/BFP markers |
| Human Improved Genome-wide Knockout CRISPR Library | Addgene: 67989 | A gRNA library against 18,010 human genes, designed for use in lentivirus. |
| GFP-BFP construct: pKLV2-U6gRNA5(gGFP)-PGKBFP2AGFP-W | Addgene: 67980 | Cas9 activity reporter with BFP and GFP. |
| Empty construct: pKLV2-U6gRNA5(empty)-PGKBFP2AGFP-W | Addgene: 67979 | Cas9 activity reporter (control) with BFP and GFP. |
Table 1: Plasmids used in this approach.
| Buffer name | Components |
| HBS (10X) | 1.5 M NaCl and 200 mM HEPES in MiliQ water, adjust to pH 7.4 |
| PBS (10X) | 80 g NaCl, 2 g KCl, 14.4 g Na2HPO4 and 2.4 g KH2PO4 in MiliQ water, adjust to pH 7.4 |
| Sodium Phosphate Buffer (80mM stock) | 7.1 g Na2HPO4.2H2O, 5.55 g NaH2PO4, adjust to pH 7.4 |
| His-purification binding buffer | 20 mM Sodium Phosphate Buffer, 0.5 M NaCl and 20 mM Imidazole, adjust to pH 7.4 |
| His-purification elution buffer | 20 mM Sodium Phosphate Buffer, 0.5M NaCl and 400 mM Imidazole, adjust to pH 7.4 |
| Diethanolamine buffer | 10% diethanolamine and 0.5 mM MgCl2 in MiliQ water, adjust to pH 9.2: |
| D10 | DMEM, 1% penicillin-streptomycin (100 units/mL) and 10% heat inactivated FBS |
Table 2: Buffers required for this study.
| Components | 10-cm dish | 6-well plate |
| 293FT cells | 70–80% confluent | 70–80% confluent |
| Transfection compatible media (Opti-MEM) (Step 5.1.2) | 3 mL | 500 µL |
| Transfection compatible media (Opti-MEM) (Step 5.1.4) | 5 mL | 2 mL |
| Lentiviral transfer vector | 3 µg | 0.5 µg |
| psPax2 (see table 1) | 7.4 µg | 1.2 µg |
| pMD2.G (see table 1) | 1.6 µg | 0.25 µg |
| PLUS reagent | 12 µL | 2 µL |
| Lipofectamine LTX | 36 µL | 6 µL |
| D10 (Step 7.1.7) | 5 mL | 1.5 mL |
| D10 (Step 7.1.8 and 7.1.10) | 8 mL | 2 mL |
Table 3: Amounts and volumes of reagents for lentivirus packaging mix.
Table 4: Primer sequences for amplifying gRNA and NGS. Please click here to view this file (Right click to download).
| Reagent | Volume per reaction | Master mix (x38) |
| Q5 Hot Start High-Fidelity 2x | 25 μL | 950 μL |
| Primer (L1/U1) mix (10 μM each) | 1 μL | 38 μL |
| Genomic DNA (1 mg/mL) | 2 μL | 72 μL |
| H2O | 22 μL | 1100 μL |
| Total | 50 μL | 1900 μL |
Table 5: PCR for the amplification of gRNAs from high complexity samples.
| Cycle number | Denature | Annealing | Extension |
| 1 | 98 °C, 30s | ||
| 2-24 | 98 °C, 10s | 61 °C, 15s | 72 °C, 20s |
| 25 | 72 °C, 2 min |
Table 6: PCR conditions for the first PCR.
| Reagent | Volume per reaction |
| KAPA HiFi HotStart ReadyMix | 25 μL |
| Primer (PE1.0/index primer) mix (5 μM each) | 2μL |
| First PCR product (40 pg/μL) | 5 μL |
| H2O | 18 μL |
| Total | 50 μL |
Table 7: PCR for the index tagging of sgRNAs from genetic screens.
| Cycle number | Denature | Annealing | Extension |
| 1 | 98 °C, 30s | ||
| 2-15 | 98 °C, 10s | 66 °C, 15s | 72 °C, 20s |
| 16 | 72 °C, 5 min |
Table 8: PCR conditions for second PCR.

Supplementary Figure S1: A guide to drawing gates for sorting the nonbinding population. (A) An ideal protein candidate for screening should have a clear shift of binding population compared to the control population and the binding should be retained on cells lacking machinery for HS biosynthesis. A heparin blocking experiment can be used in place of testing on SLC35B2 targeted cell lines. (B) Cells lacking the surface staining from the protein ectodomain but expressing BFP fluorescence from lentiviral transduction were collected. The cells displayed are from a screen for the identification of receptor for GABBR222. This figure has been modified from Sharma et al.19. Please click here to view a larger version of this figure.
{kind=link}

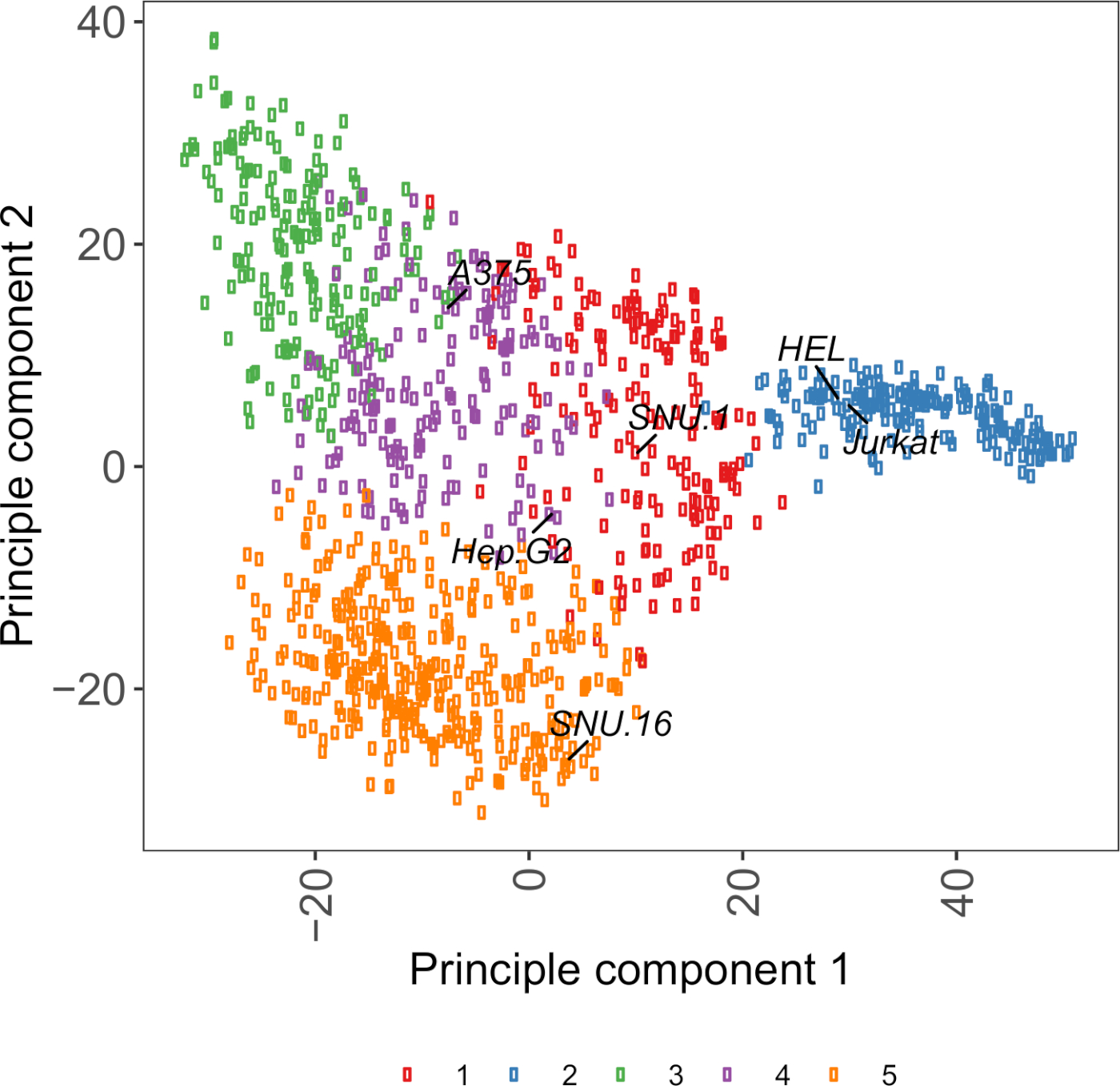
Supplementary Figure S2: Cell surface glycoprotein transcriptomics based PCA plot using RNA-seq data from over 1,000 cancer cell lines. Cell lines from Cell Model Passport27 were clustered using K-means clustering according to the FPKM values of ~1,500 cell surface glycoproteins. Representative cell lines from each cluster are labeled. Cluster 5 was entirely composed of cell lines of hematopoietic origin (also see Supplementary Table 2). Please click here to view a larger version of this figure.
{kind=link}

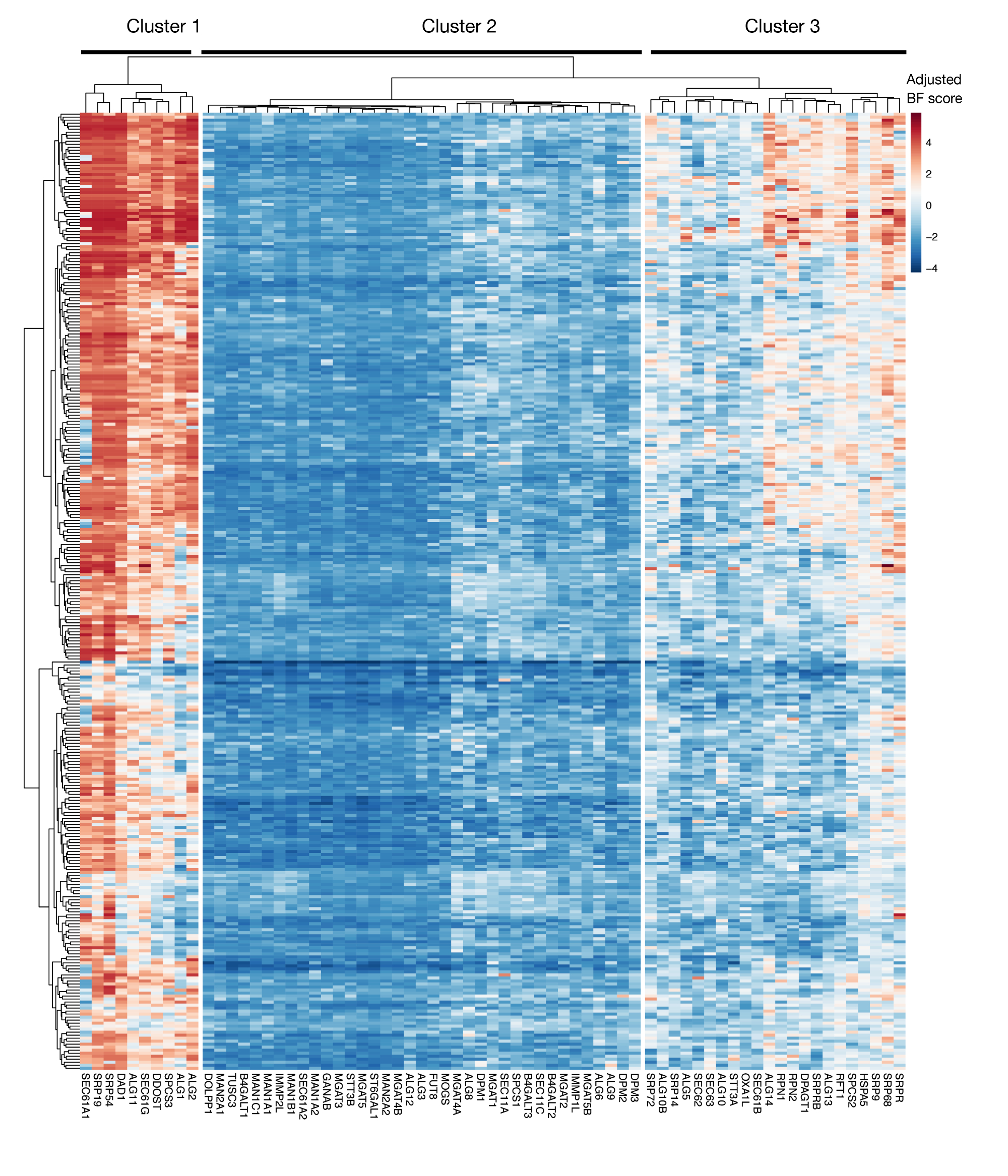
Supplementary Figure S3: Essentiality scores for KEGG-annotation protein export and N-linked glycosylation genes from project scores. Adjusted Bayes-essentiality scores for ~330 cell lines (columns, not labeled) are plotted for genes of protein export and N-linked glycosylation pathway (X-axis). Scores higher than 0 represent significant depletion in the mutant population compared to the original plasmid library. The genes can be divided into three distinct clusters that represent different levels of essentiality in the cell lines. This clustering can be used to decide the day of sorting. If the screen is performed at a late time point (day 16), it is possible that genes that are known to be essential for cells (clusters 1 and 3) will not be identified. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Table 1: Raw count files for and MAGeCK software generated gene_summary files related to the representative genetic screens. Please click here to view this file (Right click to download).
Supplementary Table 2: Clustering of cell lines according to the expression of cell surface receptors. Please click here to view this file (Right click to download).
Discussion
A CRISPR-based screening strategy to identify genes encoding cellular components involved in cellular recognition is described. A similar approach using CRISPR activation also provides a genetic alternative to identify directly interacting receptors of recombinant proteins without the need to generate large protein libraries26. However, one major advantage of this approach is that it is applicable to interactions mediated by surface molecules natively displayed on the cell and does not depend on the overexpression of receptors, which can influence the binding avidity of the receptor. Unlike other methods, therefore, this technique makes no assumptions regarding the biochemical nature or cell biology of the receptors and provides an opportunity to study interactions mediated by proteins that are normally difficult to study using biochemical approaches, such as very large proteins, or those that traverse the membrane multiple times or form complexes with other proteins, and molecules other than proteins such as glycans, glycolipids, and phospholipids. Given the genome-scale nature of the method, this approach also has the advantage of not only identifying the receptor but also additional cellular components that are required for the binding event, thereby providing insights into the cell biology of the receptor.
One of the major limitations of this method when using it to identify the receptor of an orphan protein is the initial requirement to first identify a cell line that binds to the protein. This is not always easy and identifying a cell line that displays a binding phenotype that is also permissive to genetic screens can be the time-limiting step for deploying this assay. Some cell lines tend to bind to more proteins than others. This is especially relevant for proteins that bind HS, because these proteins tend to bind to any cell line that displays HS side chains, irrespective of the native binding context. Additionally, we have observed that upregulation of syndecans (i.e., proteoglycans that contain HS) in cell lines leads to increased binding of HS-binding proteins26. This could be a factor to take into consideration when selecting the cell line for screening. However, also important to note is that the additive binding of HS does not interfere with the binding to a specific receptor. This means that if binding is observed, it is possible that it is mediated solely by HS because the binding mediated by HS in this assay is additive rather than codependent19. In such a scenario, the heparin blocking approach described can identify such behaviors without having the need to perform a full genetic screen.
A useful resource for choosing cell lines is Cell Model Passport, which contains genomics, transcriptomics, and culture condition information for ~1,000 cancer cell lines27. Depending on the biological context, cells can be chosen based on their expression profiles. To aid the selection of cell lines, we clustered ~1,000 cell lines in Cell Model Passport according to the expression of ~1,500 preannotated human cell surface glycoproteins28 (Supplementary Figure 2; cluster information for each cell line together with growth conditions are provided in Supplementary Table 2). When testing the binding of a protein with unknown function, it is useful to select a panel of representative cell lines from each cluster to increase the chance of covering a wide range of receptors. Given a choice, it is recommended to choose cell lines that are easy to culture and easy to transduce. As these cell lines will be used in genome-scale screening, it is preferable that they can be grown easily in large quantities and are permissive to lentiviral transduction, because it is the most commonly available method for delivery of sgRNA for CRISPR-based genetic screening in the later steps.
Generally, the phenotype selections are carried out in a single sort. However, this is determined by the brightness of the stained cell population compared to the control. Iterative rounds of selections could be adopted for scenarios in which the signal-to-noise ratio of the desired phenotype is low, or when the aim of the screen is to identify mutants that have strong phenotypes. When using an iterative selection approach for FACS-based screens, it is important to consider that the sorting process can cause cell death, mainly due to the sheer force of the sorter. Thus, not all collected cells will be represented in the next round of sorting.
Library complexity is a very important factor in performing successful genetic screens, especially for negative selection screens because the extent of depletion in these can only be determined by comparing results to what was present in the starting library. For negative selection screens, it is common to maintain libraries of 500-1,000 x complexity. Positive selection screens, however, are more robust to library sizes, because in such screens only a small number of mutants are expected to be selected for a particular phenotype. Therefore, in the positive selection screen described here, the library size can be decreased to 50-100x complexity without compromising the quality of the screen. In addition, in these screens it is also possible to use a control library for a given cell line on a given day as a "general control" for all samples sorted on the day for that given cell line. This will reduce the number of control libraries that need to be produced and sequenced.
Another important consideration for using this approach is the limitations of loss-of-function screens in identifying genes that are essential for in vitro cell growth. The timing of the screens is crucial in this regard, as the longer the mutant cells are kept in culture, the higher the likelihood that cells with mutations in essential genes become nonviable and are no longer represented in the mutant library. The recent genetic screens performed as a part of the Project Score initiative in over 300 cell lines show that multiple genes in the KEGG-annotated protein secretion and N-glycosylation pathway are often identified as being essential for a number of cell lines (Supplementary Figure 3)29. This can be taken into consideration when designing screens if the effect of genes required for proliferation and viability is to be investigated in the context of cellular recognition process. In this case, carrying out screens at an early timepoint (e.g., day 9 posttransduction) would be generally appropriate. However, if the approach is used to identify a few targets with strong size effects rather than general cellular pathways, it might be appropriate to perform screens at a later time point (e.g., day 15-16 posttransduction).
The results from the screening are very robust; in eight recombinant protein binding screens performed in the past, the cell surface receptor was the top hit in every case19. When using this approach to identify the interaction partner, one should therefore expect the receptor and the factors contributing to its presentation on the surface to be identified with a high statistical confidence. Once the screen is performed and a hit is validated using a single gRNA knockout, further follow-ups can be performed using existing biochemical methods such as AVEXIS4 and direct saturable binding of purified proteins using surface plasmon resonance. The approach described here is applicable for all proteins for which it is possible to generate a soluble recombinant binding probe.
In summary, this is a genome-scale CRISPR knockout approach to identify interactions mediated by cell surface proteins. This method is generally applicable to identify cellular pathways required for cell surface recognition in a wide range of different biological contexts, including between an organism's own cells (e.g., neural and immunological recognition), as well as between host cells and pathogen proteins. This method provides a genetic alternative to biochemical approaches designed for receptor identification, and because it does not require any prior assumptions regarding the biochemical nature or cell biology of the receptors it has great potential to make completely unexpected discoveries.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Wellcome Trust grant number 206194 awarded to GJW. We thank the Cytometry Core facility: Bee Ling Ng, Jennifer Graham, Sam Thompson, and Christopher Hall for help with FACS.
Materials
| Name | Company | Catalog Number | Comments |
| Anti-mouse alkaline phosphatase | Sigma | A4656 | |
| Blasticidin | Chem-Cruz | SC-204655 | |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | 13362 | |
| BSA | Sigma | A9647-100G | |
| Diethanolamine | Sigma | 398179 | |
| DMEM | Gibco | 31966-021 | |
| Dneasy Blood and Tissue kit | Qiagen | 69504 | |
| DynaMag-96 Side Magnet | Invitrogen | 12331D | |
| HEK293T packaging cells | ATCC | CRL-3216 | |
| Heparin | Sigma | H4784-1G | |
| KAPA HiFi HotStart ReadyMix | Kapa | KK2602 | |
| Lipofectamine LTX with PLUS reagent | Invitrogen | 15338100 | |
| MoFlo XDP cell sorter | BD | ||
| Ni2+-NTA agarose beads | Jena Bioscience | AC-501-25 | |
| OPTI-MEM | Life Technologies | 31985-070 | |
| OX-68 antibody | AbD Serotec | MCA1022R | |
| p-nitrophenyl phosphate | Sigma | 1040-506 | |
| PD-10 desalting columns | GE healthcare | 17085101 | |
| Polybrene | Millipore | TR-1003-G | |
| Polypropylene tubes with 5 mL bed volume | Qiagen | 34964 | |
| Proteinase K, recombinant, PCR Grade | Roche | 3115879001 | |
| Puromycin | Gibco | A11138-03 | |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | M0494L | |
| QIAquick PCR purification kit | Qiagen | 28104 | |
| SCFA filter | Nalgene | 190-2545 | |
| Sony Cell sorter | Sony | ||
| SPRI beads (Agencourt AMPure XP beads) | Beckman | A63881 | |
| Streptavidin-coated microtitre plates | Nalgene | 734-1284 | |
| Streptavidin-PE | Biolegend | 405204 |
References
- Wright, G. J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems. 5 (12), 1405-1412 (2009).
- van der Merwe, P. A., Barclay, A. N. Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends in Biochemical Sciences. 19 (9), 354-358 (1994).
- Wood, L., Wright, G. J. Approaches to identify extracellular receptor-ligand interactions. Current Opinion in Structural Biology. 56, 28-36 (2019).
- Bushell, K. M., Söllner, C., Schuster-Boeckler, B., Bateman, A., Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Research. 18 (4), 622-630 (2008).
- Visser, J. J., et al. An extracellular biochemical screen reveals that FLRTs and Unc5s mediate neuronal subtype recognition in the retina. eLife. 4, e08149(2015).
- Özkan, E., et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell. 154 (1), 228-239 (2013).
- Martinez-Martin, N., et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell. 174 (5), 1158-1171 (2018).
- Bianchi, E., Doe, B., Goulding, D., Wright, G. J. Juno is the egg Izumo receptor and is essential for mammalian fertilization. Nature. 508 (7497), 483-487 (2014).
- Mullican, S. E., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature Medicine. 23 (10), 1150-1157 (2017).
- Turner, L., et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 498 (7455), 502-505 (2013).
- Frei, A. P., et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nature Biotechnology. 30 (10), 997-1001 (2012).
- Sobotzki, N., et al. HATRIC-based identification of receptors for orphan ligands. Nature Communications. 9 (1), 1519(2018).
- Sharma, S., Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 19 (4), (2018).
- Gebre, M., Nomburg, J. L., Gewurz, B. E. CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses. 10 (2), (2018).
- Zotova, A., Zotov, I., Filatov, A., Mazurov, D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. Journal of Immunological Methods. 439, 8-14 (2016).
- Puschnik, A. S., Majzoub, K., Ooi, Y. S., Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nature Reviews. Microbiology. 15 (6), 351-364 (2017).
- Kerr, J. S., Wright, G. J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. Journal of Visualized Experiments. (61), e3881(2012).
- Tzelepis, K., et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Reports. 17 (4), 1193-1205 (2016).
- Sharma, S., Bartholdson, S. J., Couch, A. C. M., Yusa, K., Wright, G. J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Research. 28 (9), 1372-1382 (2018).
- Wang, B., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nature Protocols. 14 (3), 756-780 (2019).
- R Core team. A language and environment for statistical computing. , http://www.R-project.org (2013).
- Hart, T., et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3. 7 (8), 2719-2727 (2017).
- Kuleshov, M. V., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44 (W1), W90-W97 (2016).
- Crosnier, C., et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 480 (7378), 534-537 (2011).
- Kirk, P., et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal. 19 (15), 3896-3904 (2000).
- Chong, Z. S., Ohnishi, S., Yusa, K., Wright, G. J. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biology. 19 (1), 205(2018).
- van der Meer, D., et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Research. 47 (D1), D923-D929 (2019).
- Bausch-Fluck, D., et al. A mass spectrometric-derived cell surface protein atlas. PloS One. 10 (3), e0121314(2015).
- Behan, F. M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 568 (7753), 511-516 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved