
Method Article
Identificação do receptor de superfície celular usando telas genéticas CRISPR/Cas9 em escala de genoma
Neste Artigo
Resumo
Este manuscrito descreve uma abordagem de triagem baseada em células em escala de genoma para identificar interações receptor-ligantes extracelulares.
Resumo
A comunicação intercelular mediada por interações diretas entre receptores de superfície celular incorporados por membrana é crucial para o desenvolvimento normal e o funcionamento de organismos multicelulares. A detecção dessas interações permanece tecnicamente desafiadora, no entanto. Este manuscrito descreve uma abordagem sistemática de triagem genética de destruição em escala de genoma CRISPR/Cas9 que revela vias celulares necessárias para eventos específicos de reconhecimento da superfície celular. Este ensaio utiliza proteínas recombinantes produzidas em um sistema de expressão de proteínas mamíferas como sondas ávidas de ligação para identificar parceiros de interação em uma tela genética baseada em células. Este método pode ser usado para identificar os genes necessários para interações de superfície celular detectadas por sondas de ligação recombinantes correspondentes aos ectodomínios de receptores incorporados por membrana. É importante ressaltar que, dada a natureza em escala de genoma dessa abordagem, ele também tem a vantagem de não apenas identificar o receptor direto, mas também os componentes celulares necessários para a apresentação do receptor na superfície celular, fornecendo assim insights valiosos sobre a biologia do receptor.
Introdução
Interações extracelulares por proteínas receptoras de superfície celular direcionam processos biológicos importantes, como organização tecidual, reconhecimento de patógenos hospedeiros e regulação imunológica. Investigar essas interações é de interesse para a comunidade biomédica mais ampla, pois os receptores de membrana são alvos acionáveis de terapêuticas sistematicamente entregues, como anticorpos monoclonais. Apesar de sua importância, estudar essas interações continua sendo tecnicamente desafiador. Isso ocorre principalmente porque os receptores embutidos em membrana são anfípaticos, tornando-os difíceis de isolar das membranas biológicas para manipulação bioquímica, e suas interações são tipificadas pelas fracas afinidades de interação (KDs na faixa μM-mM)1. Consequentemente, muitos métodos comumente utilizados são inadequados para detectar essa classe de interações proteicas1,,2.
Uma série de métodos foi desenvolvida para investigar especificamente interações receptor-ligantes extracelulares que levam em consideração suas propriedades bioquímicas únicas3. Algumas dessas abordagens envolvem expressar todo o ectodomínio de um receptor como uma proteína recombinante solúvel em sistemas baseados em mamíferos ou células de insetos para garantir que essas proteínas contenham modificações pós-transicionais que são estruturalmente importantes, como glicanos e ligações de dissulfeto. Para superar a ligação de baixa afinidade, os ectodomínios são muitas vezes oligomerizados para aumentar sua avidalidade vinculante. Os ectodomínios de proteína ávida têm sido usados com sucesso como sondas de ligação para identificar parceiros de interação em telas diretas de interação proteína-proteína recombinante4,,5,,6,7. Embora métodos amplamente bem sucedidos e recombinantes à base de proteínas requerem que a ectodomina de um receptor de membrana seja produzida como uma proteína solúvel. Portanto, ele só é geralmente aplicável a proteínas que contêm uma região extracelular contígua (por exemplo, tipo de passe único I, tipo II ou ancorado em GPI) e não é geralmente adequado para complexos receptores e proteínas de membrana que abrangem a membrana várias vezes.
Técnicas de clonagem de expressão nas quais uma biblioteca de DNAs complementares (cDNAs) é transfeita em células e testada para um fenótipo de ganho de vinculação também foram usadas para identificar interações proteicas extracelulares8. A disponibilidade de grandes coleções de plasmídeos clonados e sequenciados de expressão cDNA nos últimos anos facilitou métodos nos quais linhas celulares que superexpressam cDNAs codificando receptores de superfície celular são rastreados para a ligação de proteínas recombinantes para identificar interações9,,10. As abordagens baseadas em cDNA, ao contrário dos métodos recombinantes à base de proteínas, oferecem a possibilidade de identificar interações no contexto da membrana plasmática. No entanto, o sucesso do uso de construções de expressão cDNA depende da capacidade das células de superexpressar a proteína na forma corretamente dobrada, mas isso muitas vezes requer fatores acessórios celulares, como transportadores, acompanhantes e montagem oligomerica correta. Transfetar um único cDNA pode, portanto, não ser suficiente para alcançar a expressão da superfície celular.
As técnicas de triagem que utilizam construções cDNA ou sondas de proteína recombinante são intensivas em recursos e requerem grandes coleções de bibliotecas de proteínas cDNA ou recombinante. Métodos específicos projetados baseados em espectrometria de massa foram utilizados recentemente para identificar interações extracelulares que não requerem a montagem de grandes bibliotecas. No entanto, essas técnicas requerem manipulação química da superfície celular, que pode alterar a natureza bioquímica das moléculas presentes na superfície das células e atualmente são aplicáveis apenas para interações mediadas pelas proteínas glicosiladas11,12. A maioria dos métodos disponíveis atualmente também se concentra fortemente nas interações entre proteínas, ignorando em grande parte a contribuição do microambiente da membrana, incluindo moléculas como glicanos, lipídios e colesterol.
O desenvolvimento recente de segmentação biallelelica altamente eficiente usando abordagens baseadas em CRISPR permitiu bibliotecas em escala de genoma de células sem genes definidos em uma única piscina que podem ser rastreadas de forma sistemática e imparcial para identificar componentes celulares envolvidos em diferentes contextos, incluindo dissecação de processos de sinalização celular, identificação de perturbações que conferem resistência a drogas, toxinas e patógenos, e determinação da especificidade dos anticorpos13,,14,,15,16. Aqui, descrevemos um ensaio de triagem de células eliminatórias baseadas em CRISPR em escala genoma que fornece uma alternativa às abordagens bioquímicas atuais para identificar interações receptor-ligantes extracelulares. Essa abordagem de identificar interações mediadas por receptores de membrana por telas genéticas é particularmente adequada para pesquisadores que têm um interesse focado em ligantes individuais, pois evita a necessidade de compilar grandes bibliotecas de CDNAs ou proteínas recombinantes.
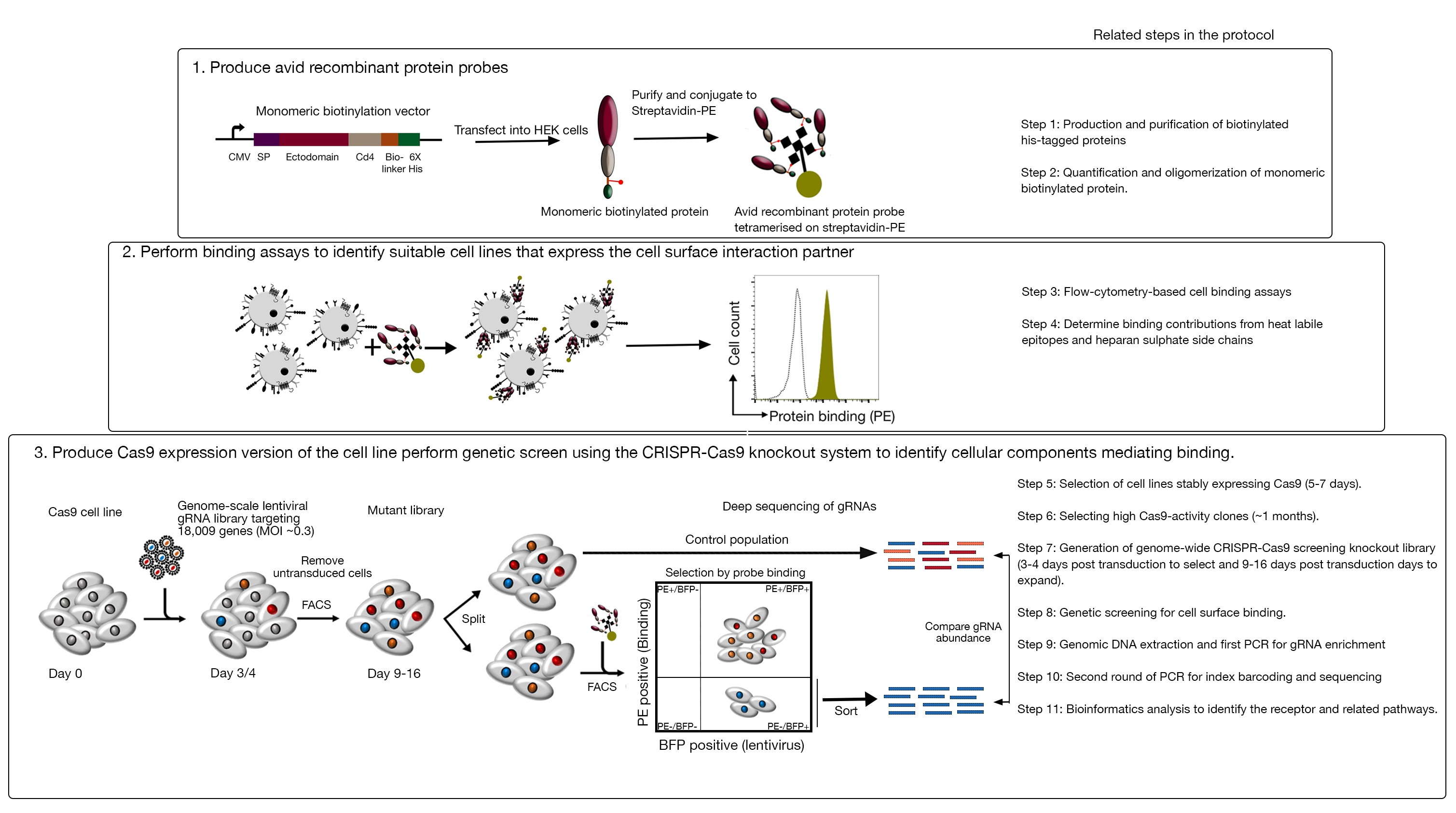
Este ensaio consiste em três passos principais: 1) Sondas de ligação de proteína recombinantes altamente ávidas que consistem nas regiões extracelulares de um receptor de interesse são produzidas e usadas em ensaios de ligação baseados em citometria de fluxo baseados em fluorescência; 2) Os ensaios de ligação são utilizados para identificar uma linha celular que expresse o parceiro de interação da sonda de proteína recombinante; 3) Uma versão expressadora cas9 da linha celular que interage com a proteína de interesse é produzida e uma tela de nocaute baseada em GENOMA CRISPR/Cas9 é realizada(Figura 1). Nesta tela genética, a ligação de uma proteína recombinante às linhas celulares é usada como um fenótipo mensurável no qual as células dentro da biblioteca de nocautes que perderam a capacidade de ligar a sonda são classificadas usando a classificação celular ativada baseada em fluorescência (FACS) e os genes que causaram a perda do fenótipo de ligação identificado pelo sequenciamento. Em princípio, os genes que codificam o receptor responsável pela ligação da ávida sonda e os necessários para sua exibição de superfície celular são identificados.
O primeiro passo deste protocolo envolve a produção de ávidas sondas proteicas recombinantes representando a ectodomina dos receptores ligados à membrana. Esses receptores são conhecidos por frequentemente manter suas funções de ligação extracelular quando seus ectodomínios são expressos como uma proteína solúvel recombinante1. Para uma proteína de interesse, proteínas recombinantes solúveis podem ser produzidas em qualquer sistema adequado de expressão de proteína eucariótica em qualquer formato, desde que possa ser oligomerizada para maior avideidade de ligação, e contém tags que podem ser usadas em ensaios de ligação baseados em fluorescência baseadas em citometria de fluxo (por exemplo, bandeira-tag, biotin-tag). Protocolos detalhados para a produção de ectodomínios solúveis de receptores de membrana utilizando o sistema de expressão proteica HEK293, bem como diferentes técnicas de multimerização e construtos de expressão proteica para a produção de proteínas pentaméricas e proteínas monoméricas foram previamente descritos1,,17. O protocolo aqui descreverá os passos para a geração de sondas ávidas fluorescentes a partir de proteínas biotinilatas monoméricas, conjugando-as a streptavidin conjugadas a um fluorocromo (por exemplo, ficoerthrina ou PE), que pode ser usado diretamente em ensaios de ligação baseados em células e tem a vantagem de não exigir um anticorpo secundário para detecção. Protocolos gerais para a realização de telas em escala de genoma já foram descritos20,,21, portanto, o protocolo se concentra principalmente nas especificidades da realização de telas de ligação de proteína recombinante baseadas em citometria de fluxo usando o sistema de triagem de nocaute CRISPR/Cas9 usando a biblioteca Humana V1 ("Yusa")18.
Protocolo
1. Produção e purificação de proteínas biotiniladas sua marcada
- Use um sistema de expressão proteica baseada em mamíferos ou células de insetos para produzir proteínas biotinínas solúveis (ver construções plasmídeos na Tabela 1).
NOTA: Um protocolo detalhado para a produção de biotina monomérica e proteínas sua marcadas usando o sistema de expressão celular HEK293 é descrito por Kerr et al.17. Proteínas ectodomínas expressas usando o sistema de expressão HEK293 são secretadas no meio da cultura. - Colete as proteínas solúveis pelo pelotização das células por centrifugação a 3.000 x g por 20 min.
- Filtre o supernante através de um filtro de 0,22 μm e adicione as contas ni2+-NTA agarose ao supernanato de proteína filtrada em uma proporção de 1:1.000 (ou seja, 50 μL de 50% de chorume agarose em 50 mL de supernante). Incubar durante a noite ou pelo menos 4-5 h a 4 °C em uma plataforma rotativa.
- Lave a coluna de polipropileno adicionando 5 mL de tampão de lavagem de sua purificação. Consulte a Tabela 2 para todas as composições tampão.
- Despeje toda a mistura de proteína de contas na coluna. Contas se acumularão na base.
- Lave as contas 2x com 15 mL de tampão de lavagem. Para evitar a diluição da proteína, retire cuidadosamente o tampão de lavagem residual da coluna com uma seringa de 5 mL e descarte.
- Adicione cuidadosamente 300-500 μL de tampão de elução de sua purificação diretamente às contas e incubar por pelo menos 1 h. Colecione a proteína eluida novamente retirando cuidadosamente o líquido usando uma seringa de 1 mL. Troque o buffer de elução para o buffer desejado (por exemplo, normalmente PBS ou HBS) usando colunas de desalar. Armazene todas as proteínas a 4 °C até que use mais.
2. Quantificação e oligomerização da proteína biotinilto monomérica
NOTA: Para aumentar a avidez vinculante, oligomerize proteínas monoméricas biotiniladas em estreptavidina tetraméricas antes de usá-las em ensaios de ligação. Alcançar as melhores relações de conjugação de proteínas monoméricas e estreptavidina-PE tetraméricas testando uma série de monômeros biotinilados contra uma concentração fixa de streptavidina e estabelecendo empiricamente a diluição mínima na qual não podem ser detectados monômeros biotinilados em excesso.
- Faça pelo menos oito diluições seriais de amostras de proteína biotinilada usando um tampão de diluição apropriado (PBS ou HBS com 1% de albumina de soro bovino [BSA]) em uma placa de 96 poços. Certifique-se de que o volume final de cada diluição seja de pelo menos 200 μL.
- Faça uma placa duplicada das amostras removendo 100 μL de cada poço e transferindo para uma nova placa de 96 poços. Inclua sempre um controle. Neste caso, os controles são proteínas somente tag (ou seja, biotininged Seu domínio Cd4 3+4 proteína). Isto será usado como uma sonda de controle em todos os ensaios de ligação.
- Diluir streptavidin-PE para 0,1 μg/mL no buffer de diluição.
- Em apenas uma das placas, adicione 100 μL do streptavidin-PE diluído. A placa duplicada servirá como controle. Adicione 100 μL de tampão de diluição na placa de controle para equalizar os volumes.
- Incubar por 20 min em temperatura ambiente (RT). Enquanto isso, bloqueie os poços de uma placa revestida de streptavidin com o tampão de diluição por 15 minutos.
- Transfira o volume total da amostra de ambas as placas para poços individuais das placas revestidas de streptavidina e incubar por 1h na RT.
- Lave a placa 3x com 200 μL de tampão de lavagem (ou seja, PBS ou HBS com 0,1% Tween-20, 2% BSA). Adicione 100 μL de 2 μg/mL de camundongo anti-rato Cd4d3+4 IgG (OX68) e incubar por 1 h no RT.
- Lave a placa 3x com o tampão de lavagem. Adicione 100 μL de um conjugado de fosfatesa alcalina anti-rato a 0,2 μg/mL por 1 h no RT.
- Lave a placa 3x com tampão de lavagem e 1x no tampão de diluição.
- Prepare fosfato p-nitrofenil a 1 mg/mL em tampão diethanolamina. Adicione 100 μL em cada poço e incubar por 15 minutos.
- Faça leituras de absorvência a 405 nm. Utilize a diluição mínima na qual não há sinal na placa como fator de diluição adequado para criar tetramers(Figura 2).
- Faça uma solução de coloração de tetramer de 10x para todas as amostras e controles incubando 4 μg/mL streptavidin-PE e a diluição proteica biotinilada apropriada por 30 minutos na RT. Armazene proteínas conjugadas em um tubo protegido pela luz a 4 °C até uso adicional.
3. Ensaios de ligação celular à base de citometria de fluxo
- Para células aderentes, remova a mídia cultural e lave 1x com PBS sem cálas divalentes. Em seguida, adicione soluções de desprendimento celular (por exemplo, EDTA). Deixe as células se soltarem por 5-10 min. Toque suavemente no frasco para liberar as células.
NOTA: Evite usar produtos à base de trippsina, pois podem cortar proteínas da superfície celular. - Colete células separadas em um tubo. Para células que crescem em suspensão (por exemplo, células HEK293), coletar diretamente as células dos frascos de cultura em um tubo.
- Células de pelotização a 200 x g por 5 min. Remova o supernaspe e resuspense a pelota no tampão de lavagem (ou seja, PBS/1% BSA).
- Conte as células usando um hemócito e ajuste a concentração para 2,5 x 105-1 x 106 células/mL. Aliquot 100 μL de mistura celular preparada em uma placa de 96 bem U ou V- com fundo. Gire a placa por 5 min a 400 x g. Remova o supernatante com uma pipeta multicanal.
- Adicione 100 μL de sondas de proteínas fluorescentes normalmente rotuladas e controles nas placas previamente preparadas com células e incubar por 1h a 4 °C. Depois de ligar por 1h, gire a placa a 400 x g por 5 min.
- Remova o supernasciente e adicione 200 μL de tampão de lavagem (ou seja, PBS/1% BSA). Misture bem por pipetting para cima e para baixo.
- Pelota as células por centrifugação a 400 x g por 5 min. Repita o passo de lavagem 1x. Depois de duas lavagens, remova completamente o supernasce e resuspenja a pelota de célula em 100 μL de PBS.
- Analise as células por citometria de fluxo. Use o laser verde-amarelo (ou seja, 561 nm) para detectar fluorescência de PE.
- Primeiro analise as células que foram manchadas com a sonda de controle. Com base na distribuição da fluorescência de PE, desenhe um portão para a população de ligação de tal forma que não mais de 1% da célula de controle caia neste portão.
- Analise a amostra e determine a fração de células que caem no portão de ligação.
NOTA: Linhas celulares que exibem uma população de ligação mais alta são desejadas para telas genéticas, pois possuem uma maior relação sinal-ruído. Idealmente, mais de 80% das células devem cair dentro deste portão.
4. Determinando contribuições vinculantes de epítopos de laboratório de calor e sidechains de sulfato heparan
NOTA: A atividade de muitas proteínas é o labile térmico, por isso a perda de atividade de ligação após o tratamento térmico é encorajadora. Aconselha-se determinar a contribuição de glicosaminoglicanos carregados negativamente, principalmente sulfato heparano (SH), na mediação da ligação das proteínas recombinantes. Isso ocorre porque a vinculação por HS no ensaio de ligação celular descrito aqui pode ser aditiva em vez de codependente em outros receptores19. Isso significa que a ligação observada pode ser totalmente mediada por cadeias laterais de HS de proteoglicas da superfície celular e não por um receptor específico. Ligar-se ao HS na superfície celular não é necessariamente inespecífico, mas sim uma propriedade de uma proteína, que é útil para saber antes de realizar uma tela genética completa.
- Prepare amostras de proteína tratadas a calor para usar em ensaios de ligação.
- Aqueça a proteína monomérica normalizada, mas não conjugada a 80 °C por 10 minutos.
- Conjugar a proteína tratada pelo calor a streptavidin-PE assumindo a mesma razão de conjugação que sua contraparte não tratada determinada por ELISA (consulte a seção 2).
- Prepare amostras de proteína bloqueadas por heparina.
- Prepare oito diluições 1:3 de heparina solúvel na PBS com concentração inicial de 2 mg/mL e volume final de 100 μL.
- Incubar 100 μL de sondas de ligação preparadas nas diluições de heparina por pelo menos 30 min.
- Use proteína tratada com calor e os 200 μL completos da mistura heparina/proteína nos ensaios de ligação descritos na seção 3. Os resultados representativos são mostrados na Figura 3A,B.
5. Seleção de linhas celulares expressando stably Cas9
NOTA: Antes que a linha celular que liga a sonda de interesse possa ser usada na triagem CRISPR, ela deve primeiro ser projetada para expressar a nuclease Cas9 e um clone altamente ativo selecionado19.
- Use o seguinte protocolo geral de produção de lentivírus para produzir lentivírus utilizando o construto lentiviral para expressão Cas9 (consulte a Tabela 1).
- Cultura HEK293-FT células em DMEM/10% FBS mídia a 37 °C e 5% CO2. Sementes HEK293-FT células 1 dia antes da transfecção de modo que eles são ~80% confluentes no dia da transfecção.
NOTA: As células HEK293FT são frouxamente aderentes; portanto, quando forem utilizados para a produção de lentivírus, considere emplacá-los em frascos de cultura revestidos com gelatina de 0,1% (w/v) para aumentar a adesão. - Faça transfecções pela manhã. Adicione vetor de transferência, mistura de embalagem e reagente de transfecção em mídia compatível com transfecção pré-armada (por exemplo, Opti-MEM). Misture invertendo o tubo 10-15x. Incubar por 5 min na RT. Consulte a Tabela 3 para obter volumes exatos.
- Adicione o reagente de transfecção conforme sugerido pelo fabricante. Misture por vórtice rápido. Incubar por 30 min na RT.
- Aspirar cuidadosamente o meio gasto. Adicione mídia compatível com transfecção à placa.
- Adicione os complexos de reagente/DNA de transfecção no lado da placa e lentamente espalhe pela placa girando muito suavemente.
- Incubar a 37 °C por 3-5 h e substituir o meio por meio D10. Incubar durante a noite.
- No dia seguinte, pela manhã, substitua o meio por um meio D10 fresco. Incubar durante a noite.
- No dia seguinte, no final da tarde, recolhe o supernatante viral. Filtrar com um filtro de 0,45 μm com baixa ligação proteica. Opcionalmente, adicione o meio D10 fresco, incubar durante a noite e lembrar o supernasal no dia seguinte.
- Os supernacantes do vírus estão estáveis a 4 °C por apenas alguns dias. Armazene a -80 °C para armazenamento a longo prazo.
NOTA: Para gerar uma preparação lentiviral altamente concentrada, que poderia ser desejável para a transdução de células difíceis de transdução, os supernacantes também podem ser concentrados por centrifugação a 6.000 x g durante a noite a 4 °C. Marque a pelota viral translúcida com uma caneta resistente ao etanol e descarte o supernante. Resuspend a pelota em 1/100 do volume original para um aumento de 100x na concentração.
- Cultura HEK293-FT células em DMEM/10% FBS mídia a 37 °C e 5% CO2. Sementes HEK293-FT células 1 dia antes da transfecção de modo que eles são ~80% confluentes no dia da transfecção.
- Transdutor as células com lentivírus.
- Placa 1 x 106 células por poço em uma placa de 6 poços com 3 mL de mídia cultural apropriada. Algumas células são mais facilmente transduzidas do que outras. Para células de fácil transdução (por exemplo, células HEK), adicione diretamente o lentivírus às células. Para células difíceis de transdução, pode ser necessário seguir um protocolo de spinoculação como descrito abaixo.
- Aliquot 2 mL de 2-5 x 106 células/mL em um tubo cônico de 15 mL.
- Adicione lentivírus junto com brometo de hexadimethrina de 8 μg/mL e incubar na RT por 30 minutos.
- Centrifugar por 100 min a 800 x g a 32 °C. Em seguida, resuspended as células na mesma mídia e adicionar a suspensão celular em frascos de cultura apropriados com mídia apropriada.
- Permitir transduções por pelo menos 24 h. Depois remova a mídia que contém o vírus e adicione um meio fresco.
- Depois de mais 24 horas, mude a mídia para uma que é complementada com os antibióticos apropriados. A construção Cas9 contém um de resistência blasticidin para seleção.
NOTA: A quantidade de blasticidina deve ser otimizada para cada linha celular realizando uma curva de morte de resposta a dose. Uma concentração de blasticidina entre 2,5-50 μg/mL deve matar a maioria das linhas celulares não traduzidas dentro de 10 dias após a transdução.
- Placa 1 x 106 células por poço em uma placa de 6 poços com 3 mL de mídia cultural apropriada. Algumas células são mais facilmente transduzidas do que outras. Para células de fácil transdução (por exemplo, células HEK), adicione diretamente o lentivírus às células. Para células difíceis de transdução, pode ser necessário seguir um protocolo de spinoculação como descrito abaixo.
- Realizar a seleção até que todas as células da placa de controle (ou seja, células não traduzidas que tenham sido tratadas com a mesma concentração de antibióticos de seleção) sejam mortas.
6. Selecionando clones de alta atividade cas9
NOTA: O Polyclonal Cas9 pode ser usado para realizar com sucesso as telas genéticas; no entanto, selecionar um clone com alta atividade Cas9 melhora os resultados de triagem18.
- Use células resistentes a blasticidina individuais de classificação única ou de célula única em poços de três placas de poços contendo mídia cultural complementada com blasticidina. Clones começarão a emergir entre 2-4 semanas. Selecione 10-20 clones e expanda em 6 placas de poço.
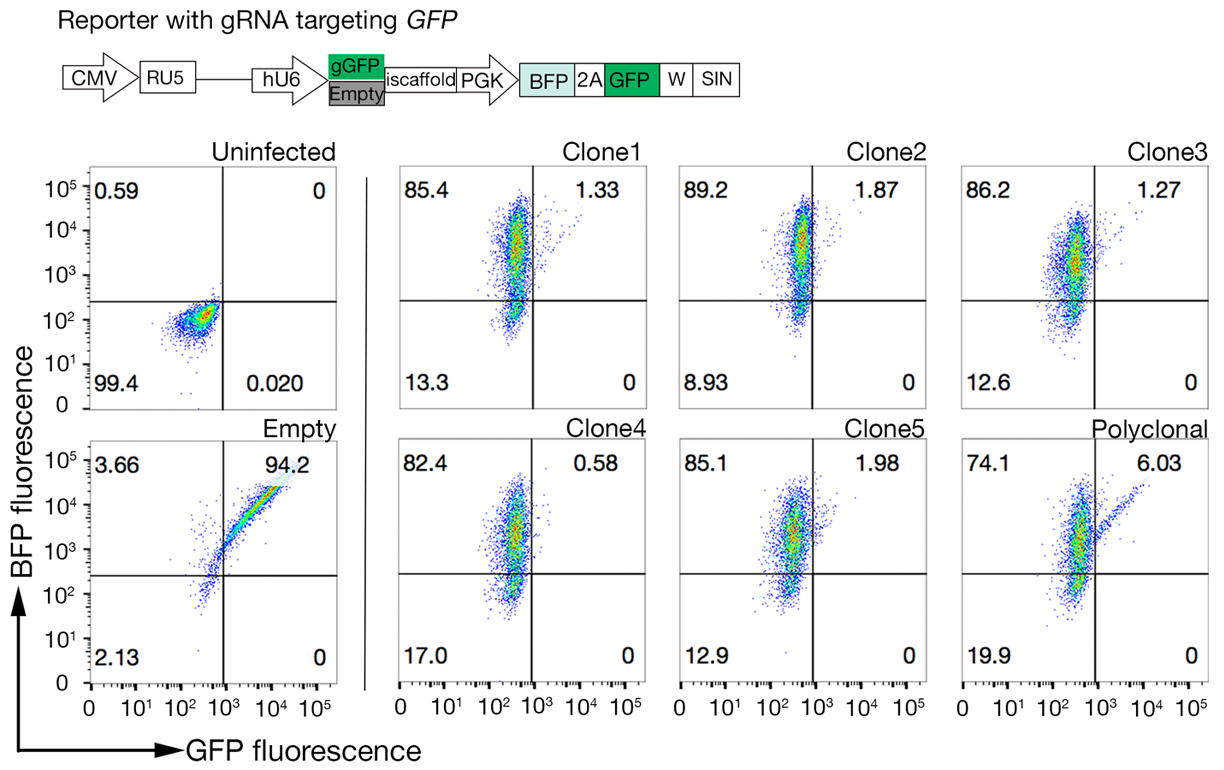
- Teste os clones para a atividade Cas9 usando o sistema GFP-BFP (proteína fluorescente verde fluorescente-proteína fluorescente azul) sistema, que usa um sistema de nocaute genético exógeno no qual as células são transduzidas ou com uma construção expressando GFP com um GFP de alvo gRNA ou um gRNA vazio como um controle18.
- Plasmids repórter de ordem: plasmídeo GFP-BFP, Plasmid Control-BFP(Tabela 1).
- Produzir lentivírus tanto para o plasmídeo GFP-BFP quanto para o plasmídeo Control-BFP utilizando o protocolo de produção de lentivírus descrito na seção 5.1.
- Transduza cada clone de linha celular que expressa Cas9 com o lentivírus codificando o sistema GFP-BFP e o Control-BFP separadamente. Siga o protocolo na seção 5.2.
- Após 3 dias de transdução, examine a fluorescência GFP-BFP de cada clone usando citometria de fluxo. Use laser de 488 nm e laser de 405 nm para detectar GFP e BFP, respectivamente.
- Quantita a atividade Cas9 em cada clone examinando a razão de BFP apenas para células positivas GFP-BFP-double. Alta atividade As células Cas9 devem ter, idealmente, >95% de eficiência de nocaute GFP(Figura 4).
7. Geração de biblioteca de nocaute de triagem CRISPR-Cas9 em todo o genoma
- Para a triagem em todo o genoma usando a biblioteca Humana V118, solicite a biblioteca em todo o genoma (consulte a Tabela 1) e prepare a biblioteca plasmida a partir da facada bacteriana usando o protocolo fornecido em "Protocolos para Replicação da Biblioteca" no manual do fabricante.
- Use a preparação plasmída da biblioteca em todo o genoma para produzir uma biblioteca lentiviral codificando gRNAs para interrupção direcionada de genes humanos usando o protocolo de produção de lentivírus descrito na seção 5.1.
NOTA: Uma boa prática é produzir um único lote de preparação lentiviral otimizado para transdução para melhorar a consistência experimental. - Use o protocolo de transdução na seção 5.2 para realizar transduções de teste em pequena escala para determinar a quantidade necessária de vírus para cada linha celular para alcançar 30% de transdução. Use a citometria de fluxo para avaliar a fluorescência BFP como proxy para eficiência de transdução.
- Para transduzir células HEK293, basta adicionar a preparação lentiviral pré-determinada a 30-50 x 106 células cultivadas em mídia de crescimento normal por ~4 h. Em seguida, remova a mídia com lentivírus e substitua por uma nova mídia de crescimento.
- Para outras linhas celulares, use o protocolo de spinocula na seção 5.2.1, mas em uma escala maior, de tal forma que um total de 30-50 x 106 células são transduzidas. Para isso, alíquota de 2 mL de 5 x 106 células/mL em um tubo cônico de 15 mL e proceda conforme indicado.
- Para linhas celulares aderentes, selecione células transduzidas adicionando puramicina 24 h após a transdução.
NOTA: Otimize as concentrações de puromicina realizando uma curva de morte de resposta a dose. Normalmente, as concentrações entre 1-10 μg/mL devem matar células não traduzidas dentro de 3-5 dias. Evite usar concentrações mais altas de puromicina porque isso pode aumentar as chances de selecionar células que foram transduzidas por mais de um guia de RNA (sgRNA). - Para células de suspensão, colher células transduzidas (ou seja, BFP positivo) 3 dias após a transferência usando um classificador celular e gerar bibliotecas que contenham pelo menos 10 x 106 células. Uma vez selecionado usando BFP, cresça as células em mídia suplementada com quantidade adequada de puramicina.
NOTA: Evite seleções apenas com puromicina para linhas de células suspensas, pois é difícil remover células mortas e detritos de culturas celulares suspensas que podem interferir na classificação celular. - Biblioteca mutante cultural para 9-16 dias pós-tradução com passagem regular a cada 2-3 dias.
8. Triagem genética para ligação de superfície celular
- Pelota a biblioteca celular mutante a 200 x g por 5 min e resuspend as células na PBS.
- Divida as células em dois tubos cônicos de 15 mL com pelo menos 50 x 106 células em cada tubo.
- Gire um tubo cônico a 200 x g por 5 min, remova o sobrenante e congele a pelota da célula a -20 °C. Esta é a população de controle e será processada mais tarde.
- Resuspende a pelota no outro tubo em 10 mL de PBS/1% BSA. Reserve 100 μL de células como um controle negativo em uma placa de poço de 96.
- Adicione a proteína recombinante pré-conconteante adequada à suspensão celular no tubo cônico e proteínas de controle negativas à placa de poço 96.
- Realize a coloração celular por pelo menos 1 h a 4 °C em um rotor de bancada com rotação suave (6 rpm).
- Pelota as células a 200 x g por 5 min, remova o supernasce. Realize duas etapas de lavagem e, em seguida, resuspenja as células em 5 mL de PBS.
- Coe as células através de um coador de células de 30 μm para remover aglomerados celulares. Analise usando um classificador de fluxo.
- Use a amostra de controle negativo para portar células BFP+/PE.
- Classifique a amostra e colete as células BFP+/PE. Os portões de classificação dependerão da ligação das células à proteína, mas normalmente são coletadas 1-5% das amostras negativas de PE. Um exemplo de portão de classificação é fornecido na Figura Suplementar 1.
- Colete 500.000-1.000.000 células do portão selecionado. Dado o baixo número de células, considere coletar as amostras em um tubo de centrifugação de 1,5 mL para minimizar as perdas.
- Pelota as células classificadas por centrifugação a 500 x g por 5 min. Remova cuidadosamente o sobrenante e descarte. É possível armazenar a pelota a -20 °C por até 6 meses.
9. Extração genômica de DNA e primeiro PCR para enriquecimento de gRNA
- Extrair DNA genômico da população de controle de alta complexidade.
- Se a população de controle foi congelada a -20 °C, tire o tubo cônico e adicione PBS. Mantenha o gelo para descongelar a pelota.
- Use um kit comercial (ver Tabela de Materiais) usando as recomendações do fabricante para extrair DNA genômico de 50 x 106 células. Ajuste a concentração de DNA para 1 mg/mL.
- Para cada amostra, configure uma mistura mestra para PCR correspondente a 72 μg de DNA. Aliquot 50 μL por poço em 36 poços de uma placa PCR de 96 poços. As sequências de primer necessárias estão listadas na Tabela 4. Use o guia na Tabela 5 e 6.
- Resolva 5 μL do PCR de 6-12 amostras representativas em um gel de agarose de 2% (w/v). Uma única banda clara a ~250 bp deve ser observada. Se as bandas estiverem fracas, repita o PCR para mais 2-3 ciclos.
- Use uma pipeta multicanal para coletar 5 μL de produtos PCR de cada poço (180 μL no total) e acumulá-los em um reservatório com 900 μL de tampão de ligação de um kit comercial (ver Tabela de Materiais).
- Purifique os produtos PCR usando um kit comercial de purificação pcr. DNA eluto em 50 μL de tampão de elução de um kit comercial (ver Tabela de Materiais) e medir a concentração de DNA.
- Amostras que foram classificadas para a perda de fenótipo de ligação são improváveis de serem compostas de um grande número de clones independentes. Portanto, não é necessário realizar PCR com 72 μg de DNA. Isole o DNA usando um kit comercial apropriado (ver Tabela de Materiais). Configure reações 3-4 pcr usando o protocolo descrito antes (seção 9.1.3) com 100 ng/μL DNA. Se o número de células classificadas for inferior a 100.000 usam lises celulares em vez de preparações genômicas de DNA.
- Alíquota de aproximadamente 10.000 células/poço em uma placa PCR de 96 poços.
- Pelota as células na placa e remova cuidadosamente a maior parte do supernatante. A pelota não será visível.
- Adicione 25 μL de água em cada poço e aqueça as amostras a 95 °C por 10 minutos.
- Adicione 5 μL de 2 mg/mL de proteína recém-diluída K a cada poço por 1 h e incubar a 56 °C. Em seguida, aqueça a amostra por 10 min a 95 °C para inativar a proteína K.
- Use 10 μL de mistura de liseto celular por reação PCR. Os lysates devem ser usados dentro de 24 horas.
10. Segunda rodada de PCR para codificação e sequenciamento de índices
- Diluir os produtos da primeira rodada pcr para 40 pg/μL.
- Configure um PCR por amostra (use o guia fornecido nas Tabelas 7 e 8). O uso de uma polimerase de alta fidelidade é importante para minimizar os erros introduzidos pela polimerase durante a amplificação do sgRNA.
- Resolva 5 μL de produtos PCR em um gel de agarose de 2% (p/v). Uma única banda clara a ~330 bps deve ser observada.
- Purifique os produtos PCR usando contas paramagnéticas adicionando 31,5 μL de (volume total de 0,7x) de contas resuspended aos produtos PCR, misturando bem e incubando por 5 min na RT.
- Coloque o tubo em um rack magnético por 3 minutos. As contas devem ser capturadas na lateral da placa e a solução deve ser clara. Remova cuidadosamente e descarte o supernaspe.
- Adicione 150 μl de 80% de etanol recém-preparado ao tubo. Incubar por 30 s e, em seguida, remover cuidadosamente e descartar o supernaspe.
- Repita o passo 13.6, desta vez com 180 μL. Em seguida, seque as contas por 5 minutos.
- Remova o tubo do ímã. Alvo de DNA elute de contas em 35 μL de tampão EB estéril. Incubar por 3 minutos, depois coloque o tubo de volta no rack magnético por 3 minutos.
- Transfira aproximadamente 30 μL do supernascimento contendo os produtos PCR elucidos para um tubo limpo.
- Sequenciar as amostras em uma plataforma de sequenciamento de próxima geração. Para a biblioteca humanv1 gRNA, use o primer personalizado listado na Tabela 4 para sequenciar 19 bp.
11. Análise bioinformática para identificar o receptor e caminhos relacionados
- Mapear sequências de população classificada e não sortida para a biblioteca de referência usando a função de contagem de MAGeCK. A função renderá um arquivo de contagem bruta(Tabela Complementar 1).
NOTA: Instruções detalhadas sobre a instalação do MAGeCK e o uso de diferentes funções dentro do MAGeCK são descritas em um protocolo publicado anteriormente por Wang et al.20. - Verifique o padrão técnico da biblioteca de controle usada na tela.
- Normalizar a contagem bruta e usar o pacote ggplot2 em R21 ou software equivalente para traçar um gráfico de função de densidade cumulativa empírica das contagens em plasmídeos e controlar amostras não sortidas(Figura 5A).
- Execute a função de teste do MAGeCK usando contagens da população plasmida como "controle" e as contagens de amostras de controle não variadas como a amostra de "teste". A função é produzir um arquivo de resumo genético(Tabela Suplementar 1).
- Abra o arquivo de resumo genético e desenhe a distribuição de alterações de dobra de log (colunaneg|lfc) para genes essenciais e não essenciais categorizados anteriormente22 (Figura 5B).
- Selecione genes significativamente esgotados(neg|fdr < 0,05) e realize a análise de enriquecimento da via usando o pacote enrichador23 ou quaisquer pacotes de enriquecimento de via equivalente em R(Figura 5C).
- Execute a função de teste do MAGeCK com a configuração padrão. Use contagens brutas de amostra de controle não variada como "controle" e conta a partir de amostra classificada como "tratamento" ao realizar a análise.
- Abra o arquivo de resumo genético gerado pelo MAGeCK e classifique a coluna pos|rank em ordem ascendente. Use FDR ( colunapos|fdr) < 0,05 como ponto de corte para identificação de acertos. O receptor é geralmente classificado altamente, muitas vezes na primeira posição.
- Plote as pontuações do Robust-Ranking-Algorithm (RRA) para seleção positiva(pos|score) em R ou um software equivalente(Figura 5D).
- Selecione os hits genéticos e realize a análise de enriquecimento da via usando o pacote de enriquecimento ou quaisquer pacotes de enriquecimento de vias equivalentes em R para identificar as vias enriquecidas.
Resultados
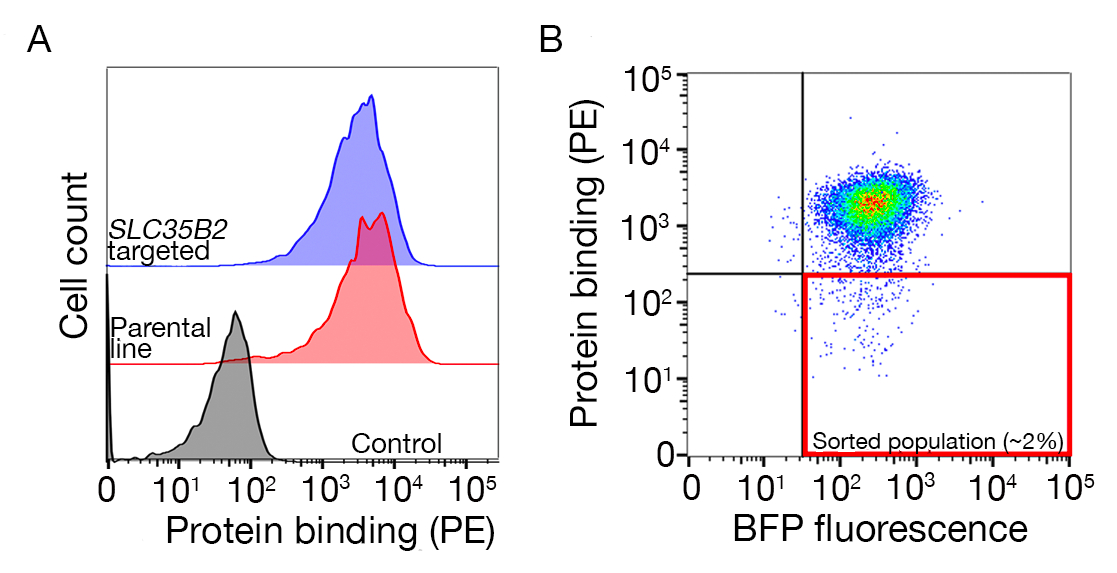
São fornecidos dados de sequenciamento de duas telas de knockout representativas em escala de genoma para a identificação do parceiro vinculante do TNFSF9 e P. falciparum RH5 realizados nas células NCI-SNU-1 e HEK293 , respectivamente (Tabela Suplementar 1). O comportamento de ligação do RH5 foi afetado tanto pelo sulfato heparano quanto pelo seu conhecido receptor BSG24 (Figura 3C),enquanto o TNFRSF9 especificamente ligado ao seu receptor conhecido TNFSF9 e não perdeu a vinculação após a pré-insubtação com heparina solúvel. A proteína 3 na Figura 3B representa tnfrsf9.
Para ambas as linhas celulares, também estão previstas a distribuição de gRNAs na biblioteca mutante de controle após 3 dias (9, 14 e 16 dias após a transferência)(Tabela Suplementar 1). A distribuição do gRNA revelou que a complexidade da biblioteca foi mantida ao longo do experimento (Figura 5A). A tela genética para identificação do ligante para TNFSF9 foi realizada no dia 14 pós-transmissão, enquanto que para RH5 foi realizada dia 9 pós-transmissão. A qualidade técnica das telas foi avaliada examinando a distribuição de dobras observadas de gRNAs visando um conjunto de referência de genes não essenciais em comparação com a distribuição para conjunto de referência de genes essenciais22 (Figura 5B). Além disso, o enriquecimento no nível da via também revelou que as vias essenciais esperadas foram identificadas e significativamente enriquecidas na população "desistência" ao comparar a amostra de controle com a biblioteca plasmida original. Um exemplo com a amostra NCI-SNU-1 do dia 14 é retratado na Figura 5C.
A distribuição dos gRNAs no controle versus população classificada usando a função de teste de MAGeCK (ver Tabela Suplementar 1 para a saída de resumo genético de MAGeCK) foi usada para identificar o receptor a partir das telas fenotípicas. O escore RRA modificado relatado pelo MAGeCK na análise de nível genético é plotado em relação aos genes classificados pelos valores p. A pontuação rra no MAGeCK fornece uma medida na qual os gRNAs são classificados consistentemente acima do esperado. Na tela para TNFRSF9, o topo foi O TNFSF9, que é um parceiro de ligação conhecido da TNFRSF9 (Figura 5D). Além disso, também foram identificados vários genes relacionados à via TP53. No caso do RH5, além do receptor conhecido (BSG)e do gene necessário para a produção dos GAGs sulfatos(SLC35B2),também foi identificado um gene adicional(SLC16A1)(Figura 5E). SLC16A1 é um acompanhante necessário para o tráfico de BSG à superfície das células25. Juntos, esses resultados demonstram a capacidade da tela de identificar receptores interagindo diretamente e os componentes celulares necessários para que esse receptor seja expresso na superfície das células de forma funcional.

Figura 1: Visão geral da abordagem de triagem genética para identificar receptores de superfície celular. Este ensaio consiste em três grandes passos: Primeiro, proteínas recombinantes representando a ectodomínia dos receptores de superfície celular são expressas em uma linha celular que pode adicionar modificações pós-transacionais estruturalmente críticas, como as células HEK293. Proteína monomédica ectodominas são oligomerizadas conjugando-se ao streptavidin-PE para aumentar sua avidalidade vinculante. Em segundo lugar, essas ávidas sondas são usadas em ensaios de ligação celular onde manchas brilhantes nas linhas celulares indicadas por uma mudança proeminente na fluorescência de PE (em verde) em comparação com uma proteína de controle negativo (em preto) demonstra a presença de um parceiro de ligação de superfície celular. Em terceiro lugar, as linhas celulares cas9-positivas do receptor são selecionadas e a triagem em escala de genoma usando gRNAs visando a grande maioria dos genes codificadores de proteínas é realizada. Ao gerar bibliotecas mutantes, é comum usar 30% de eficiência de transdução, que é baseada na probabilidade de distribuição de Poisson que garante que cada célula receba um único gRNA de tal forma que o fenótipo resultante seja atribuído a um nocaute específico. O marcador BFP expresso pelas células transduzidas é usado para selecionar células contendo gRNAs usando FACS. As telas fenotípicas são realizadas entre 9 e 16 dias após a transferência. No dia da tela, a população total de células mutantes é dividida em duas. Uma metade é mantida como a população de controle e a outra metade é selecionada para ligação recombinante de proteínas. As células da biblioteca mutante que não são mais capazes de ligar a proteína recombinante são classificadas usando FACS e o enriquecimento de gRNAs na população classificada versus controle é usado para identificar genes necessários para a ligação da superfície celular da sonda ávida rotulada. São indicadas etapas do protocolo que requerem tempo considerável. Este número foi modificado a partir de Sharma et al.19. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Estabelecendo as proporções de proteína biotinilada para streptavidina-PE utilizando um método baseado em ELISA. Um exemplo de estratégia de conjugação streptavidin-PE usada para gerar uma ávida sonda a partir de uma proteína monomérica biotinilada. Uma série de diluição de monômeros biotinilados foi incubada contra uma concentração fixa de streptavidina. A diluição mínima na qual não podem ser detectados monômeros biotinilados em excesso foram determinados pela ELISA. A ELISA foi realizada com ou sem pré-insubação de uma série de diluições proteicas com 10 ng de streptavidin-PE. Na presença de streptavidin-PE, a diluição mínima na qual não foi identificado nenhum sinal (preto circulado) e a quantidade de proteína necessária para a saturação foi calculada para gerar uma solução de estoque de 10x com 4 μg/mL streptavidin-PE. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Vinculação representativa de proteínas às linhas celulares. (A) A ligação proteica às linhas celulares teve um claro aumento da fluorescência associada às células em comparação com a amostra de controle. O tratamento térmico (80 °C por 10 min) de proteína recombinante revogou toda a ligação a um controle negativo, demonstrando que o comportamento de ligação dependia de proteínas corretamente dobradas. (B) Diferentes classes de comportamento de ligação proteica às superfícies celulares; dependência de GAGs. Da esquerda para a direita, as proteínas podem ser classificadas em três tipos: proteína tipo 1 apenas adsorbs para HS. Essas proteínas perdem sua ligação após a pré-insubtação com concentrações de heparina acima de 0,2 mg/mL. Proteína tipo 2 liga-se ao HS, além de um receptor específico. Essas proteínas perdem a vinculação parcial nos experimentos de pré-bloqueio. Proteína tipo 3 não liga HS. Essas proteínas não perdem a vinculação em comparação com as linhas parentais. (C) Um exemplo de proteína (ou seja, RH5) que se liga ao SS e a um receptor específico de forma aditiva. Direcionar o receptor (ou seja, BSG) ou enzimas necessárias para a síntese de HS (por exemplo, SLC35B2, EXTL3) apenas reduz parcialmente a vinculação do RH5 às células relativas aos controles. Linhas policlonais transduzidas podem ser usadas em tais experimentos para estabelecer comportamentos vinculativos. Este número foi modificado a partir de Sharma et al.19. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Selecionando linhas de células clonais com alta atividade Cas9. A eficiência de edição de genomas de linhas policlonais e clonadas de linhas celulares NCI-SNU-1 foi avaliada usando o sistema de repórter GFP-BFP, no qual as linhas celulares foram transduzidas com vírus com um GFP codificado por gRNA ou sem (ou seja, "vazio"). Um esquema é retratado. A citometria de fluxo foi utilizada para testar tanto a expressão BFP quanto a GFP após a transdução e comparada ao controle não infectado. A expressão GFP foi utilizada como proxy para a atividade Cas9, enquanto a expressão BFP marcou células transduzidas. O perfil de células infectadas não infectadas e vazias parecia semelhante para todos os clones. Perfis representativos são retratados no painel esquerdo. Todos os cinco clones da linha celular NCI-SNU-1 apresentaram maior perda de GFP em comparação com a linha policlonal (painel direito), com o clone 4 mostrando a maior eficiência com a menor população refratária. Este número foi modificado a partir de Sharma et al.19. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Resultados representativos de telas genéticas para a identificação dos parceiros de ligação da superfície celular. (A) Parcelas de função de distribuição cumulativa comparando a abundância de gRNA na biblioteca plasmida com as bibliotecas mutantes das células HEK-293-E e NCI-SNU-1 nos dias 9, 14 e 16 dias pós-transmissão. Para qualquer número, a função de densidade cumulativa relata a porcentagem de pontos de dados abaixo desse limite. A pequena mudança da população celular mutante em comparação com a população plasmida original representa o esgotamento em um subconjunto de gRNAs em comparação com a biblioteca plasmida. (B) Distribuição de alterações de dobra de log em genes previamente categorizados como essenciais (vermelho) ou não essenciais (preto) nas linhas celulares HEK293 e NCI-SNU-1. A distribuição de dobras para genes não essenciais centrados em ~0, enquanto que para genes essenciais mudou para a esquerda para mudanças negativas de dobra. (C) Caminhos significativamente enriquecidos em genes esgotados na população de controle mutante NCI-SNU-1 14 dias após a transferência. Foram identificadas vias conhecidas de células-essenciais. (D) Robust Rank Algorithm (RRA)-score para genes que foram enriquecidos nas células classificadas que perderam a capacidade de ligar a sonda TNFRSF9. Os genes foram classificados de acordo com a pontuação da RRA. O conhecido parceiro de interação TNFSF9 e genes relacionados à via TP53 (rotulado em vermelho) foram identificados na tela. (E) E E E-scores de RRA ordenados para genes identificados a partir da análise de enriquecimento gRNA necessário para a vinculação RH5 às células HEK293 (painel esquerdo). SLC35B2 e SLC16A1 foram identificados dentro de um limiar de taxa de falsa descoberta (FDR) de 5%. Dois genes adicionais na via de biossíntese de HS (ou seja, EXTL3 e NDST1) foram identificados dentro da FDR de 25%. Esquema representando o caminho geral da biossíntese gag com os genes relevantes mapeados para as etapas correspondentes (painel 2). Os genes necessários para o compromisso com a biogênese sulfato de condroitina (ou seja, CSGALNACT1/2) não foram identificados na tela. Este número foi modificado a partir de Sharma et al.19. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Nome plasmídeo | Plasmídeo # | Usar |
| Construção de expressão proteica: CD200RCD4d3+4-bio-linker-his | Addgene: 36153 | Produção de proteína recombinante com CD4d3+4, biotina e 6-suas tags. |
| pMD2.G | Addgene: 12259 | Envelope VSV-G expressando plasmid; produção de lentivírus |
| psPAX2 | Addgene: 12260 | Plasmid de embalagens lentivirais, produção de lentivírus |
| Construção cas9: pKLV2-EF1a-Cas9Bsd-W | Addgene: 68343 | Produção de linha Cas9 expressando constitutivamente |
| construção de expressão gRNA: pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W | Addgene: 67974 | Vetor de expressão crispr gRNA com um andaime melhorado e marcadores puro/BFP |
| Biblioteca de CRISPR de genoma melhorado humano | Addgene: 67989 | Uma biblioteca de gRNA contra 18.010 genes humanos, projetada para uso em lentivírus. |
| Construção GFP-BFP: pKLV2-U6gRNA5(gGFP)-PGKBFP2AGFP-W | Addgene: 67980 | Repórter de atividades Cas9 com BFP e GFP. |
| Construção vazia: pKLV2-U6gRNA5(vazio)-PGKBFP2AGFP-W | Addgene: 67979 | Repórter de atividade Cas9 (controle) com BFP e GFP. |
Tabela 1: Plasmids utilizados nesta abordagem.
| Nome tampão | Componentes |
| HBS (10X) | 1,5 M NaCl e 200 mM HEPES em água MiliQ, ajuste para pH 7.4 |
| PBS (10X) | 80 g NaCl, 2 g KCl, 14,4 g Na2HPO4 e 2,4 g KH2PO4 em água MiliQ, ajuste para pH 7.4 |
| Tampão fosfato de sódio (estoque de 80mM) | 7.1 g Na2HPO4.2H2O, 5,55 g NaH2PO4,ajuste ao pH 7.4 |
| Tampão de ligação de purificação dele | Tampão fosfato de sódio de 20 mM, 0,5 M NaCl e 20 mM Imidazol, ajuste ao pH 7.4 |
| Tampão de elução de sua purificação | Tampão fosfato de sódio de 20 mM, 0,5M NaCl e 400 mM Imidazol, ajuste ao pH 7.4 |
| Tampão diethanolamina | 10% diethanolamina e 0,5 mM MgCl2 na água MiliQ, ajuste ao pH 9.2: |
| D10 | DMEM, 1% penicilina-estreptomicina (100 unidades/mL) e 10% de FBS inativados |
Tabela 2: Tampões necessários para este estudo.
| Componentes | Prato de 10 cm | Placa de 6 poços |
| Células de 293FT | 70-80% confluente | 70-80% confluente |
| Mídia compatível com transfecção (Opti-MEM) (Passo 5.1.2) | 3 mL | 500 μL |
| Mídia compatível com transfecção (Opti-MEM) (Passo 5.1.4) | 5 mL | 2 mL |
| Vetor de transferência lentiviral | 3 μg | 0,5 μg |
| psPax2 (ver tabela 1) | 7,4 μg | 1,2 μg |
| pMD2.G (ver tabela 1) | 1,6 μg | 0,25 μg |
| Reagente PLUS | 12 μL | 2 μL |
| Lipofectamina LTX | 36 μL | 6 μL |
| D10 (Passo 7.1.7) | 5 mL | 1,5 mL |
| D10 (Passo 7.1.8 e 7.1.10) | 8 mL | 2 mL |
Tabela 3: Quantidades e volumes de reagentes para mistura de embalagens de lentivírus.
Tabela 4: Sequências de primer para amplificar gRNA e NGS. Clique aqui para ver este arquivo (clique com o botão direito do mouse para baixar).
| Reagente | Volume por reação | Mix mestre (x38) |
| Q5 Hot Start High-Fidelity 2x | 25 μL | 950 μL |
| Mistura de primer (L1/U1) (10 μM cada) | 1 μL | 38 μL |
| DNA genômico (1 mg/mL) | 2 μL | 72 μL |
| H2O | 22 μL | 1100 μL |
| Total | 50 μL | 1900 μL |
Tabela 5: PCR para a amplificação de gRNAs de amostras de alta complexidade.
| Número do ciclo | Desnaturar | Recozimento | Extensão |
| 1 | 98 °C, 30s | ||
| 2-24 | 98 °C, 10s | 61 °C, 15s | 72 °C, 20s |
| 25 | 72 °C, 2 min |
Tabela 6: Condições de PCR para o primeiro PCR.
| Reagente | Volume por reação |
| KAPA HiFi HotStart ReadyMix | 25 μL |
| Mix de primer (PE1.0/index primer) (5 μM cada) | 2μL |
| Primeiro produto PCR (40 pg/μL) | 5 μL |
| H2O | 18 μL |
| Total | 50 μL |
Tabela 7: PCR para a marcação de índice de sgRNAs a partir de telas genéticas.
| Número do ciclo | Desnaturar | Recozimento | Extensão |
| 1 | 98 °C, 30s | ||
| 2-15 | 98 °C, 10s | 66 °C, 15s | 72 °C, 20s |
| 16 | 72 °C, 5 min. |
Tabela 8: Condições de PCR para o segundo PCR.

Figura Suplementar S1: Um guia para desenhar portões para classificação da população não vinculante. (A) Um candidato ideal à proteína para triagem deve ter uma clara mudança de população vinculante em comparação com a população de controle e a vinculação deve ser retida em células sem maquinário para a biossíntese de HS. Um experimento de bloqueio de heparina pode ser usado no lugar de testes em linhas celulares direcionadas ao SLC35B2. (B) Foram coletadas células sem a coloração superficial da proteína ectodomína, mas expressando fluorescência de BFP a partir da transdução lentiviral. As células exibidas são de uma tela para identificação do receptor para GABBR222. Este número foi modificado a partir de Sharma et al.19. Clique aqui para ver uma versão maior desta figura.
{kind=link}

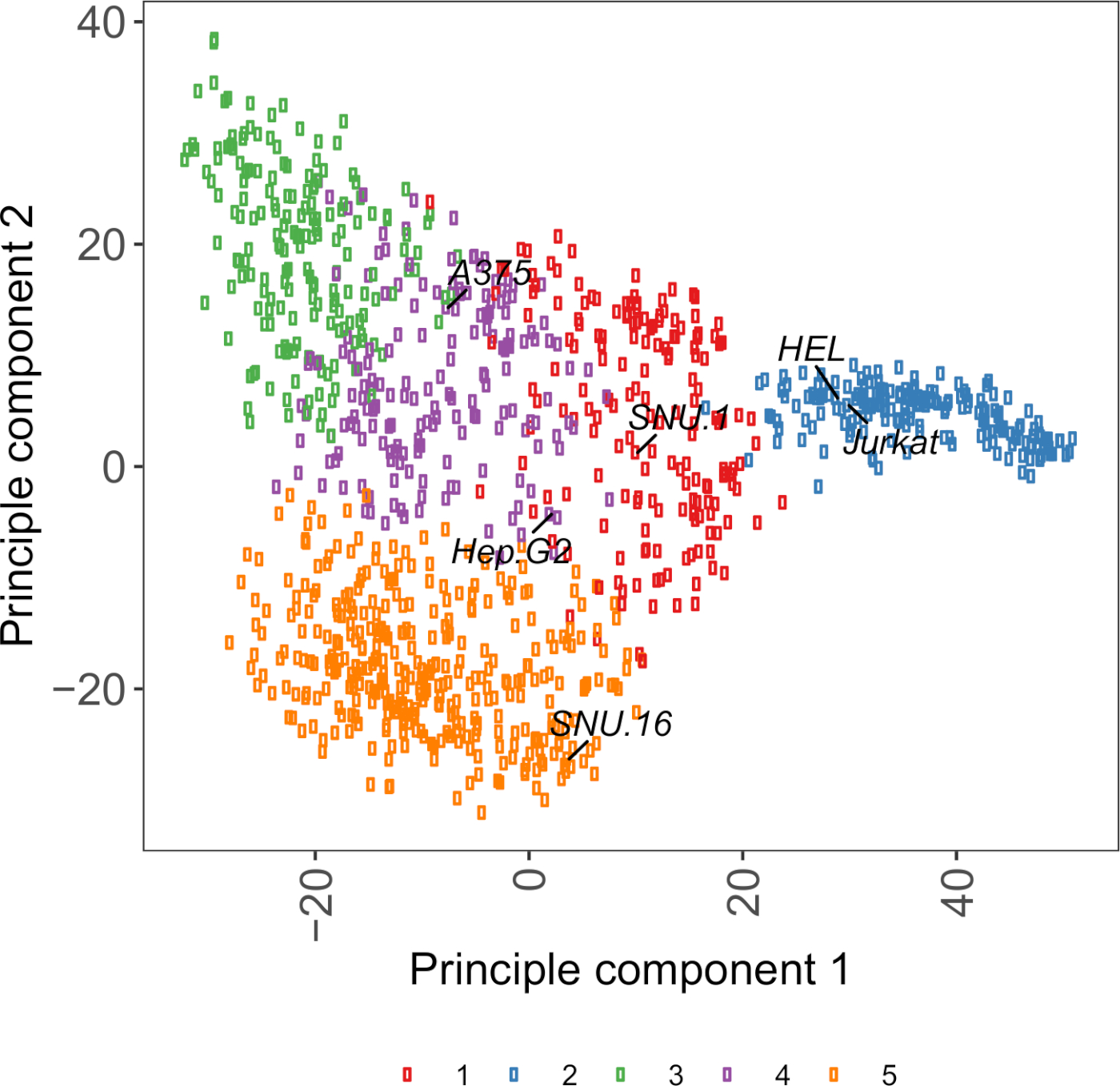
Figura suplementar S2: Transcrição de glicoproteína de superfície celular baseada em plotagem PCA usando dados RNA-seq de mais de 1.000 linhas de células cancerosas. As linhas celulares do Cell Model Passport27 foram agrupadas usando agrupamento de meios K de acordo com os valores FPKM de ~1.500 glicoproteínas de superfície celular. As linhas de células representativas de cada cluster são rotuladas. O cluster 5 era inteiramente composto de linhas celulares de origem hematopoiética (também ver Tabela Suplementar 2). Clique aqui para ver uma versão maior desta figura.
{kind=link}

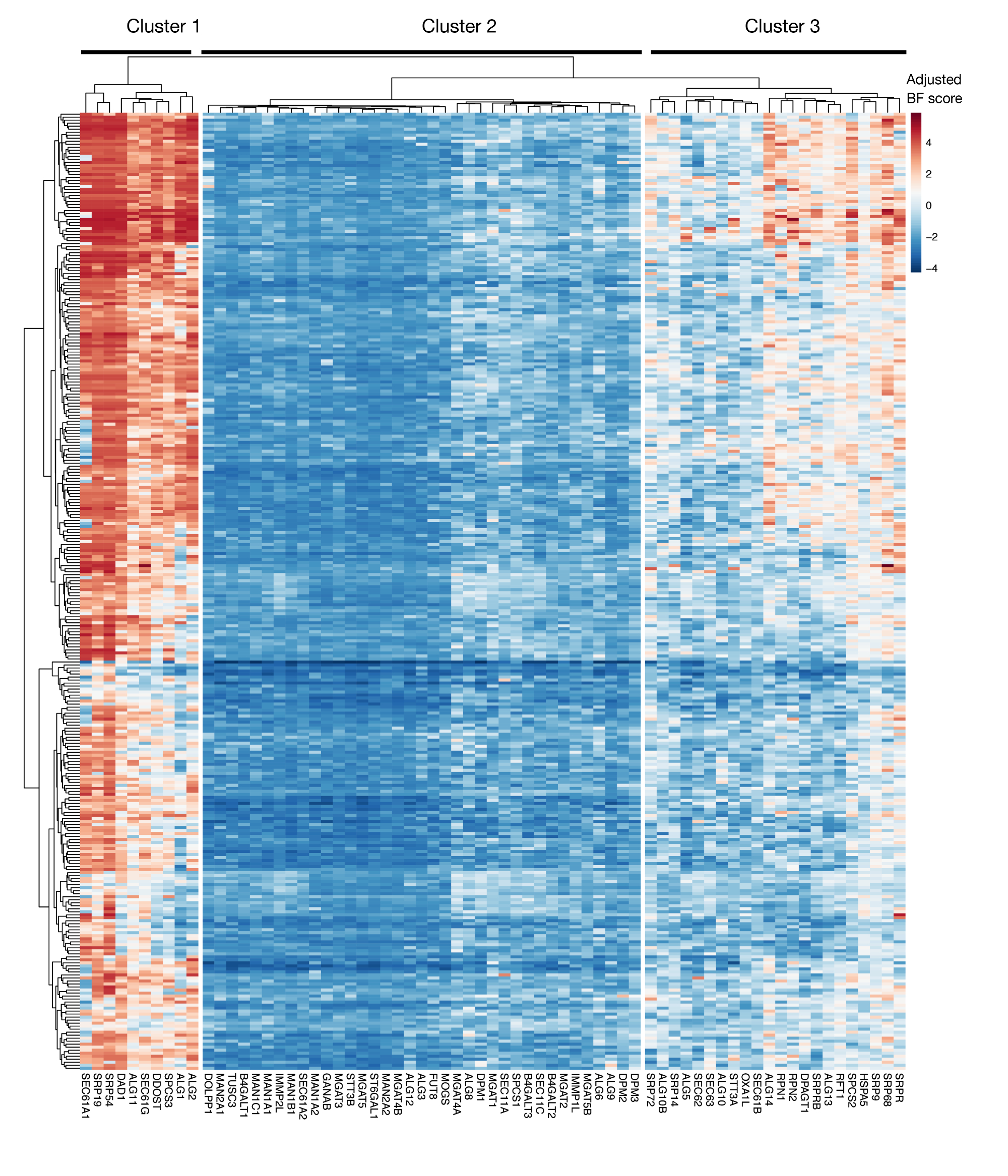
Figura suplementar S3: Pontuações de essencialidade para exportação de proteínas de anotação kegg e genes de glicossylation ligados a N a partir de escores de projeto. Os escores de essencialidade de Bayes ajustados para ~330 linhas celulares (colunas, não rotuladas) são plotados para genes de exportação de proteínas e via de glicosylation ligada a N (eixo X). Pontuações acima de 0 representam um esgotamento significativo na população mutante em comparação com a biblioteca plasmida original. Os genes podem ser divididos em três aglomerados distintos que representam diferentes níveis de essencialidade nas linhas celulares. Este agrupamento pode ser usado para decidir o dia da classificação. Se a tela for realizada em um ponto de tempo tardio (dia 16), é possível que genes que são conhecidos como essenciais para as células (clusters 1 e 3) não sejam identificados. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela suplementar 1: Arquivos de contagem bruta para e software MAGeCK gerados gene_summary arquivos relacionados às telas genéticas representativas. Clique aqui para ver este arquivo (clique com o botão direito do mouse para baixar).
Tabela suplementar 2: Agrupamento de linhas celulares de acordo com a expressão dos receptores de superfície celular. Clique aqui para ver este arquivo (clique com o botão direito do mouse para baixar).
Discussão
Uma estratégia de triagem baseada em CRISPR para identificar genes codificando componentes celulares envolvidos no reconhecimento celular é descrita. Uma abordagem semelhante usando a ativação do CRISPR também fornece uma alternativa genética para identificar receptores diretamente interagindo de proteínas recombinantes sem a necessidade de gerar grandes bibliotecas proteicas26. No entanto, uma grande vantagem dessa abordagem é que ela é aplicável às interações mediadas por moléculas superficiais nativamente exibidas na célula e não depende da superexpressão dos receptores, o que pode influenciar a avidez vinculante do receptor. Ao contrário de outros métodos, portanto, essa técnica não faz suposições sobre a natureza bioquímica ou biologia celular dos receptores e oferece uma oportunidade de estudar interações mediadas por proteínas que normalmente são difíceis de estudar usando abordagens bioquímicas, como proteínas muito grandes, ou aquelas que atravessam a membrana várias vezes ou formam complexos com outras proteínas, e moléculas diferentes de proteínas como glicanos, glicólipídios e fosfolipídios. Dada a natureza em escala genoma do método, essa abordagem também tem a vantagem de não apenas identificar o receptor, mas também componentes celulares adicionais que são necessários para o evento de ligação, fornecendo assim insights sobre a biologia celular do receptor.
Uma das principais limitações deste método ao usá-lo para identificar o receptor de uma proteína órfã é o requisito inicial para identificar primeiro uma linha celular que se liga à proteína. Isso nem sempre é fácil e identificar uma linha celular que exibe um fenótipo de ligação que também é permissivo às telas genéticas pode ser o passo que limita o tempo para a implantação deste ensaio. Algumas linhas celulares tendem a se ligar a mais proteínas do que outras. Isso é especialmente relevante para proteínas que ligam HS, porque essas proteínas tendem a se ligar a qualquer linha celular que exiba cadeias laterais de HS, independentemente do contexto de ligação nativa. Além disso, observamos que a regulação dos sindecanos (ou seja, proteoglycans que contêm HS) nas linhas celulares leva ao aumento da vinculação das proteínas de ligação de SH26. Isso pode ser um fator a ser levado em consideração ao selecionar a linha celular para triagem. No entanto, também é importante notar que a ligação aditiva do HS não interfere na vinculação a um receptor específico. Isso significa que, se a vinculação for observada, é possível que seja mediada unicamente pelo SS porque a vinculação mediada pelo HS neste ensaio é aditiva e não codependente19. Nesse cenário, a abordagem de bloqueio de heparina descrita pode identificar tais comportamentos sem ter a necessidade de realizar uma tela genética completa.
Um recurso útil para a escolha de linhas celulares é o Cell Model Passport, que contém informações genômicas, transcriômicas e condições de cultura para ~1.000 linhas de células cancerígenas27. Dependendo do contexto biológico, as células podem ser escolhidas com base em seus perfis de expressão. Para auxiliar na seleção de linhas celulares, agrupamos ~1.000 linhas celulares no Cell Model Passport de acordo com a expressão de ~1.500 glicoproteínas de superfície de células humanas pré-anotadas28 (Figura Suplementar 2; informações de cluster para cada linha celular juntamente com condições de crescimento são fornecidas na Tabela Suplementar 2). Ao testar a ligação de uma proteína com função desconhecida, é útil selecionar um painel de linhas celulares representativas de cada cluster para aumentar a chance de cobrir uma ampla gama de receptores. Dada a escolha, recomenda-se escolher linhas celulares fáceis de cultivar e fáceis de transduzir. Como essas linhas celulares serão usadas na triagem em escala de genoma, é preferível que elas possam ser cultivadas facilmente em grandes quantidades e sejam permissivas à transdução lentiviral, pois é o método mais comumente disponível para a entrega de sgRNA para triagem genética baseada em CRISPR nas etapas posteriores.
Geralmente, as seleções de fenótipo são realizadas em um único tipo. No entanto, isso é determinado pelo brilho da população celular manchada em comparação com o controle. Rodadas iterativas de seleções poderiam ser adotadas para cenários em que a relação sinal-ruído do fenótipo desejado é baixa, ou quando o objetivo da tela é identificar mutantes que têm fenótipos fortes. Ao usar uma abordagem de seleção iterativa para telas baseadas em FACS, é importante considerar que o processo de classificação pode causar morte celular, principalmente devido à força do classificador. Assim, nem todas as células coletadas serão representadas na próxima rodada de triagem.
A complexidade da biblioteca é um fator muito importante na realização de telas genéticas bem sucedidas, especialmente para telas de seleção negativas, pois a extensão do esgotamento nelas só pode ser determinada comparando resultados com o que estava presente na biblioteca inicial. Para telas de seleção negativas, é comum manter bibliotecas de 500-1.000 x de complexidade. Telas de seleção positivas, no entanto, são mais robustas para tamanhos de biblioteca, porque nessas telas apenas um pequeno número de mutantes são esperados para ser selecionado para um fenótipo específico. Portanto, na tela de seleção positiva descrita aqui, o tamanho da biblioteca pode ser reduzido para 50-100x de complexidade sem comprometer a qualidade da tela. Além disso, nessas telas também é possível usar uma biblioteca de controle para uma determinada linha celular em um determinado dia como um "controle geral" para todas as amostras classificadas no dia para aquela linha celular dada. Isso reduzirá o número de bibliotecas de controle que precisam ser produzidas e sequenciadas.
Outra consideração importante para o uso dessa abordagem são as limitações das telas de perda de função na identificação de genes essenciais para o crescimento celular in vitro. O tempo das telas é crucial nesse sentido, pois quanto mais tempo as células mutantes são mantidas na cultura, maior a probabilidade de que células com mutações em genes essenciais se tornem inviáveis e não sejam mais representadas na biblioteca mutante. As recentes telas genéticas realizadas como parte da iniciativa Project Score em mais de 300 linhas celulares mostram que múltiplos genes na secreção de proteínas anotadas pelo KEGG e na via de glicossylação N-glicossylation são frequentemente identificados como essenciais para uma série de linhas celulares (Figura Suplementar 3)29. Isso pode ser levado em consideração ao projetar telas se o efeito dos genes necessários para a proliferação e viabilidade deve ser investigado no contexto do processo de reconhecimento celular. Neste caso, a realização de telas em um ponto de tempo antecipado (por exemplo, dia 9 pós-tradução) seria geralmente apropriada. No entanto, se a abordagem for usada para identificar alguns alvos com efeitos de tamanho forte em vez de vias celulares gerais, pode ser apropriado executar telas em um ponto de tempo posterior (por exemplo, dia 15-16 pós-tradução).
Os resultados da triagem são muito robustos; em oito telas de ligação de proteína recombinantes realizadas no passado, o receptor de superfície celular foi o principal atingido em cada caso19. Ao utilizar essa abordagem para identificar o parceiro de interação, deve-se, portanto, esperar que o receptor e os fatores que contribuem para sua apresentação na superfície sejam identificados com alta confiança estatística. Uma vez que a tela é realizada e um hit é validado usando um único nocaute de gRNA, outros acompanhamentos podem ser realizados usando métodos bioquímicos existentes, como AVEXIS4 e ligação satural direta de proteínas purificadas usando ressonância de plasmon superficial. A abordagem aqui descrita é aplicável para todas as proteínas para as quais é possível gerar uma sonda de ligação recombinante solúvel.
Em resumo, esta é uma abordagem de nocaute CRISPR em escala de genoma para identificar interações mediadas por proteínas da superfície celular. Este método é geralmente aplicável para identificar vias celulares necessárias para o reconhecimento da superfície celular em uma ampla gama de diferentes contextos biológicos, incluindo entre as células próprias de um organismo (por exemplo, reconhecimento neural e imunológico), bem como entre células hospedeiras e proteínas patógenas. Este método fornece uma alternativa genética às abordagens bioquímicas projetadas para identificação de receptores, e por não exigir quaisquer suposições prévias sobre a natureza bioquímica ou biologia celular dos receptores, tem grande potencial para fazer descobertas completamente inesperadas.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este trabalho foi apoiado pela bolsa wellcome trust número 206194 concedida à GJW. Agradecemos à instalação do Núcleo de Citometria: Bee Ling Ng, Jennifer Graham, Sam Thompson e Christopher Hall por ajuda com a FACS.
Materiais
| Name | Company | Catalog Number | Comments |
| Anti-mouse alkaline phosphatase | Sigma | A4656 | |
| Blasticidin | Chem-Cruz | SC-204655 | |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | 13362 | |
| BSA | Sigma | A9647-100G | |
| Diethanolamine | Sigma | 398179 | |
| DMEM | Gibco | 31966-021 | |
| Dneasy Blood and Tissue kit | Qiagen | 69504 | |
| DynaMag-96 Side Magnet | Invitrogen | 12331D | |
| HEK293T packaging cells | ATCC | CRL-3216 | |
| Heparin | Sigma | H4784-1G | |
| KAPA HiFi HotStart ReadyMix | Kapa | KK2602 | |
| Lipofectamine LTX with PLUS reagent | Invitrogen | 15338100 | |
| MoFlo XDP cell sorter | BD | ||
| Ni2+-NTA agarose beads | Jena Bioscience | AC-501-25 | |
| OPTI-MEM | Life Technologies | 31985-070 | |
| OX-68 antibody | AbD Serotec | MCA1022R | |
| p-nitrophenyl phosphate | Sigma | 1040-506 | |
| PD-10 desalting columns | GE healthcare | 17085101 | |
| Polybrene | Millipore | TR-1003-G | |
| Polypropylene tubes with 5 mL bed volume | Qiagen | 34964 | |
| Proteinase K, recombinant, PCR Grade | Roche | 3115879001 | |
| Puromycin | Gibco | A11138-03 | |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | M0494L | |
| QIAquick PCR purification kit | Qiagen | 28104 | |
| SCFA filter | Nalgene | 190-2545 | |
| Sony Cell sorter | Sony | ||
| SPRI beads (Agencourt AMPure XP beads) | Beckman | A63881 | |
| Streptavidin-coated microtitre plates | Nalgene | 734-1284 | |
| Streptavidin-PE | Biolegend | 405204 |
Referências
- Wright, G. J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems. 5 (12), 1405-1412 (2009).
- van der Merwe, P. A., Barclay, A. N. Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends in Biochemical Sciences. 19 (9), 354-358 (1994).
- Wood, L., Wright, G. J. Approaches to identify extracellular receptor-ligand interactions. Current Opinion in Structural Biology. 56, 28-36 (2019).
- Bushell, K. M., Söllner, C., Schuster-Boeckler, B., Bateman, A., Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Research. 18 (4), 622-630 (2008).
- Visser, J. J., et al. An extracellular biochemical screen reveals that FLRTs and Unc5s mediate neuronal subtype recognition in the retina. eLife. 4, e08149 (2015).
- Özkan, E., et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell. 154 (1), 228-239 (2013).
- Martinez-Martin, N., et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell. 174 (5), 1158-1171 (2018).
- Bianchi, E., Doe, B., Goulding, D., Wright, G. J. Juno is the egg Izumo receptor and is essential for mammalian fertilization. Nature. 508 (7497), 483-487 (2014).
- Mullican, S. E., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature Medicine. 23 (10), 1150-1157 (2017).
- Turner, L., et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 498 (7455), 502-505 (2013).
- Frei, A. P., et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nature Biotechnology. 30 (10), 997-1001 (2012).
- Sobotzki, N., et al. HATRIC-based identification of receptors for orphan ligands. Nature Communications. 9 (1), 1519 (2018).
- Sharma, S., Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 19 (4), (2018).
- Gebre, M., Nomburg, J. L., Gewurz, B. E. CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses. 10 (2), (2018).
- Zotova, A., Zotov, I., Filatov, A., Mazurov, D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. Journal of Immunological Methods. 439, 8-14 (2016).
- Puschnik, A. S., Majzoub, K., Ooi, Y. S., Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nature Reviews. Microbiology. 15 (6), 351-364 (2017).
- Kerr, J. S., Wright, G. J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. Journal of Visualized Experiments. (61), e3881 (2012).
- Tzelepis, K., et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Reports. 17 (4), 1193-1205 (2016).
- Sharma, S., Bartholdson, S. J., Couch, A. C. M., Yusa, K., Wright, G. J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Research. 28 (9), 1372-1382 (2018).
- Wang, B., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nature Protocols. 14 (3), 756-780 (2019).
- Hart, T., et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3. 7 (8), 2719-2727 (2017).
- Kuleshov, M. V., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44 (W1), W90-W97 (2016).
- Crosnier, C., et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 480 (7378), 534-537 (2011).
- Kirk, P., et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal. 19 (15), 3896-3904 (2000).
- Chong, Z. S., Ohnishi, S., Yusa, K., Wright, G. J. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biology. 19 (1), 205 (2018).
- van der Meer, D., et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Research. 47 (D1), D923-D929 (2019).
- Bausch-Fluck, D., et al. A mass spectrometric-derived cell surface protein atlas. PloS One. 10 (3), e0121314 (2015).
- Behan, F. M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 568 (7753), 511-516 (2019).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados