
Method Article
Identifizierung von Zelloberflächenrezeptoren mit Genom-Scale CRISPR/Cas9 Genetic Screens
In diesem Artikel
Zusammenfassung
Dieses Manuskript beschreibt einen zellbasierten Screening-Ansatz im Genommaßstab zur Identifizierung extrazellulärer Rezeptor-Ligand-Wechselwirkungen.
Zusammenfassung
Die interzelluläre Kommunikation, die durch direkte Wechselwirkungen zwischen membraneingebetteten Zelloberflächenrezeptoren vermittelt wird, ist entscheidend für die normale Entwicklung und Funktionsweise multizellulärer Organismen. Die Erkennung dieser Wechselwirkungen bleibt jedoch technisch anspruchsvoll. Dieses Manuskript beschreibt einen systematischen CRISPR/Cas9-Knockout-Genscreening-Ansatz im Genommaßstab, der zelluläre Pfade aufzeigt, die für bestimmte Zelloberflächenerkennungsereignisse erforderlich sind. Dieser Test nutzt rekombinante Proteine, die in einem Säugetierprotein-Expressionssystem produziert werden, als aviane Bindungssonden, um Interaktionspartner in einem zellbasierten genetischen Screen zu identifizieren. Diese Methode kann verwendet werden, um die Gene zu identifizieren, die für Zelloberflächeninteraktionen notwendig sind, die von rekombinanten Bindungssonden nachgewiesen werden, die den Ektodomänen von Membran-eingebetteten Rezeptoren entsprechen. Wichtig ist, dass dieser Ansatz angesichts des genomskaligen Charakters dieses Ansatzes nicht nur den direkten Rezeptor identifiziert, sondern auch die zellulären Komponenten, die für die Darstellung des Rezeptors an der Zelloberfläche benötigt werden, und somit wertvolle Einblicke in die Biologie des Rezeptors liefert.
Einleitung
Extrazelluläre Wechselwirkungen durch Zelloberflächenrezeptorproteine leiten wichtige biologische Prozesse wie Gewebeorganisation, Wirtspathogenerkennung und Immunregulation. Die Untersuchung dieser Wechselwirkungen ist für die breitere biomedizinische Gemeinschaft von Interesse, da Membranrezeptoren umsetzbare Ziele systematisch gelieferter Therapeutika wie monoklonale Antikörper sind. Trotz ihrer Bedeutung bleibt das Studium dieser Wechselwirkungen technisch anspruchsvoll. Dies liegt vor allem daran, dass membranintegrierte Rezeptoren amphipathisch sind, was sie schwierig macht, von biologischen Membranen für biochemische Manipulation zu isolieren, und ihre Wechselwirkungen werden durch die schwachen Wechselwirkungen(KDs im M-mM-Bereich)1typisiert. Folglich sind viele häufig verwendete Methoden ungeeignet, um diese Klasse von Proteinwechselwirkungen zu erkennen1,2.
Eine Reihe von Methoden wurde entwickelt, um speziell extrazelluläre Rezeptor-Ligand-Wechselwirkungen zu untersuchen, die ihre einzigartigen biochemischen Eigenschaften berücksichtigen3. Eine Reihe dieser Ansätze beinhaltet die Expression der gesamten Ektodomäne eines Rezeptors als lösliches rekombinantes Protein in Säugetier- oder Insektenzellsystemen, um sicherzustellen, dass diese Proteine posttranslationale Modifikationen enthalten, die strukturell wichtig sind, wie Glykane und Disulfidbindungen. Um die Bindung mit geringer Affinität zu überwinden, werden die Ectodomains oft oligomerisiert, um ihre Bindungssucht zu erhöhen. Avid Protein-Ectodomains wurden erfolgreich als Bindungssonden eingesetzt, um Interaktionspartner in direkten rekombinanten Protein-Protein-Interaktionsbildschirmen4,5,6,7zu identifizieren. Obwohl im Großen und Ganzen erfolgreich, erfordern rekombinante proteinbasierte Methoden, dass die Ectodomain eines Membranrezeptors als lösliches Protein produziert wird. Daher ist es nur allgemein anwendbar auf Proteine, die eine zusammenhängende extrazelluläre Region enthalten (z. B. Single-Pass Typ I, Typ II oder GPI-verankert) und ist im Allgemeinen nicht für Rezeptorkomplexe und Membranproteine geeignet, die die Membran mehrfach überspannen.
Expressionsklonierungstechniken, bei denen eine Bibliothek komplementärer DNAs (cDNAs) in Zellen transfiziert und auf einen Gain-of-Binding-Phänotyp getestet wurde, wurden auch verwendet, um extrazelluläre Protein-Protein-Wechselwirkungen zu identifizieren8. Die Verfügbarkeit großer Sammlungen von geklonten und sequenzierten cDNA-Expressionsplasmiden in den letzten Jahren hat Methoden erleichtert, bei denen Zelllinien, die cDNAs kodieren, zellzelloberflächenrezeptoren kodiert, auf die Bindung rekombinanter Proteine untersucht werden, um Wechselwirkungen9,10zu identifizieren. Die cDNA-Überexpressions-basierten Ansätze bieten im Gegensatz zu rekombinanten proteinbasierten Methoden die Möglichkeit, Wechselwirkungen im Kontext der Plasmamembran zu identifizieren. Der Erfolg der Verwendung von cDNA-Expressionskonstrukten hängt jedoch von der Fähigkeit der Zellen ab, das Protein in der korrekt gefalteten Form zu überexprimieren, aber dies erfordert oft zelluläre Zubehörfaktoren wie Transporter, Chaperones und korrekte oligomere Baugruppe. Die Transfikierung einer einzelnen cDNA reicht daher möglicherweise nicht aus, um eine Zelloberflächenexpression zu erreichen.
Screening-Techniken mit cDNA-Konstrukten oder rekombinanten Proteinsonden sind ressourcenintensiv und erfordern große Sammlungen von cDNA- oder rekombinanten Proteinbibliotheken. Speziell entwickelte Massenspektrometrie-basierte Methoden wurden in letzter Zeit verwendet, um extrazelluläre Interaktionen zu identifizieren, die nicht die Zusammenstellung großer Bibliotheken erfordern. Diese Techniken erfordern jedoch eine chemische Manipulation der Zelloberfläche, die die biochemische Natur der moleküle verändern kann, die auf der Oberfläche der Zellen vorhanden sind, und derzeit nur für Wechselwirkungen anwendbar sind, die durch glykosylierte Proteine vermittelt werden11,12. Die meisten der derzeit verfügbaren Methoden konzentrieren sich auch stark auf die Wechselwirkungen zwischen Proteinen, während weitgehend ignoriert den Beitrag der Membran Mikroumgebung, einschließlich Moleküle wie Glykane, Lipide, und Cholesterin.
Die jüngste Entwicklung eines hocheffizienten bialleleischen Targetings mit CRISPR-basierten Ansätzen hat es ermöglicht, genombasierte Bibliotheken von Zellen ohne definierte Gene in einem einzigen Pool zu identifizieren, die systematisch und unvoreingenommen untersucht werden können, um zelluläre Komponenten zu identifizieren, die in verschiedenen Kontexten beteiligt sind, einschließlich der Zerlegung zellulärer Signalisierungsprozesse, der Identifizierung von Störungen, die Resistenzen gegen Medikamente, Toxine und Krankheitserreger verleihen, und der Bestimmung der Spezifizität von Antikörpern13,14,, 16 ,,1616. Hier beschreiben wir einen CRISPR-basierten Knockout-Zell-Screening-Assay im Genommaßstab, der eine Alternative zu den aktuellen biochemischen Ansätzen zur Identifizierung extrazellulärer Rezeptor-Liganden-Wechselwirkungen bietet. Dieser Ansatz zur Identifizierung von Wechselwirkungen, die von Membranrezeptoren durch genetische Screens vermittelt werden, eignet sich besonders für Forscher, die ein konzentriertes Interesse an einzelnen Liganden haben, da sie die Notwendigkeit vermeidet, große Bibliotheken von cDNAs oder rekombinanten Proteinen zusammenzustellen.
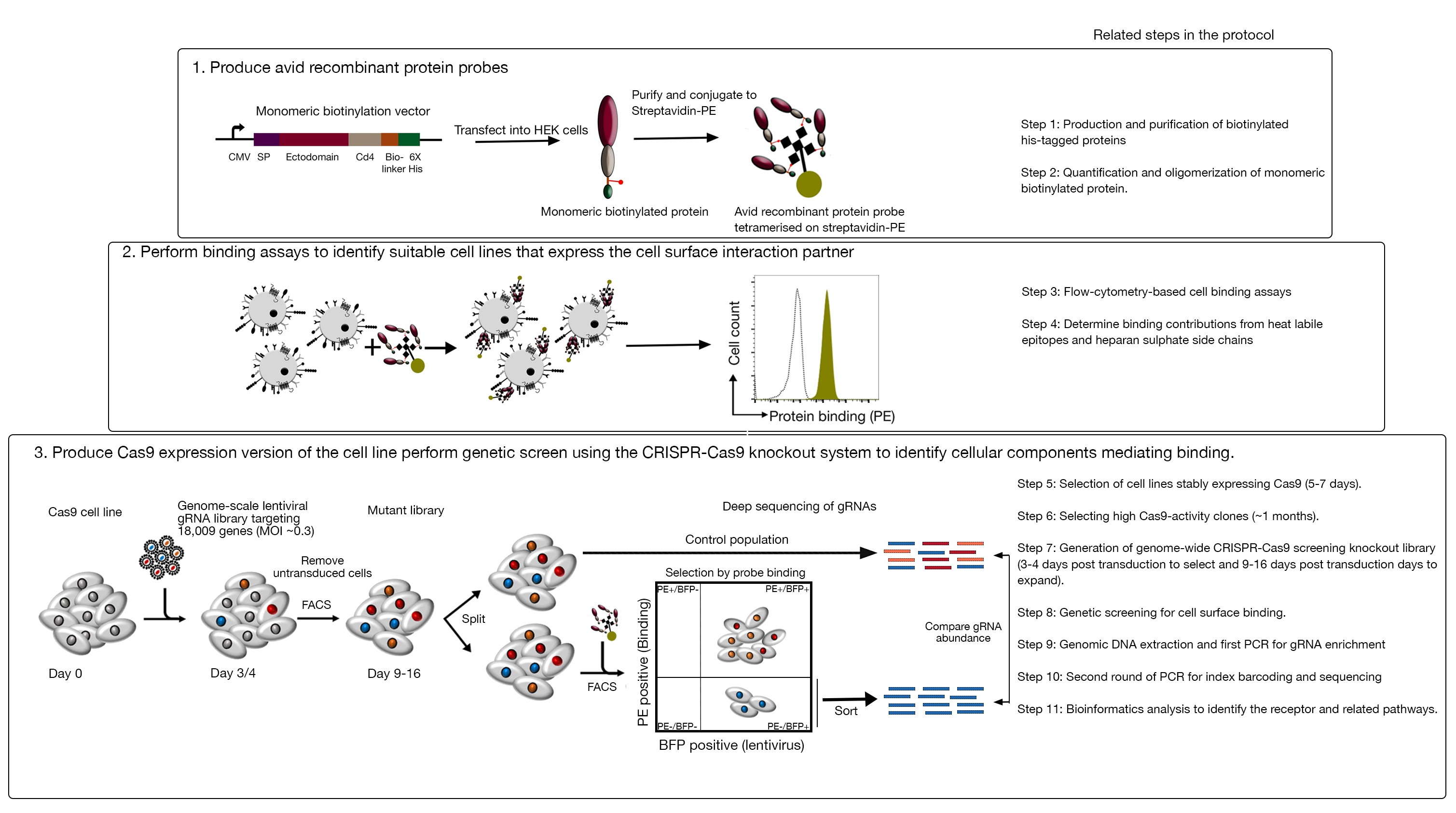
Dieser Test besteht aus drei Hauptschritten: 1) Hochgradig aviante rekombinante Proteinbindungssonden, die aus den extrazellulären Regionen eines Rezeptors von Interesse bestehen, werden produziert und in fluoreszenzbasierten Flusszytometrie-basierten Bindungstests verwendet; 2) Die Bindungstests werden verwendet, um eine Zelllinie zu identifizieren, die den Interaktionspartner der rekombinanten Proteinsonde ausdrückt; 3) Es wird eine Cas9-exättige Version der Zelllinie erzeugt, die mit dem Protein von Interesse interagiert, und es wird ein CRISPR/Cas9-basierter Knockout-Bildschirm im Genommaßstab durchgeführt (Abbildung 1). In diesem genetischen Screen wird die Bindung eines rekombinanten Proteins an Zelllinien als messbarer Phänotyp verwendet, bei dem Zellen innerhalb der Knockout-Bibliothek, die die Fähigkeit verloren haben, die Sonde zu binden, mit fluoreszenzbasierter aktivierter Zellsortierung (FACS) und den Genen sortiert werden, die den Verlust des durch Sequenzierung identifizierten Bindungsphänotyps verursacht haben. Grundsätzlich werden die Gene identifiziert, die für den Rezeptor kodieren, der für die Bindung der Avianssonde verantwortlich ist, und die Gene, die für die Zelloberflächenanzeige erforderlich sind.
Der erste Schritt dieses Protokolls beinhaltet die Produktion von avianen rekombinanten Proteinsonden, die die Ectodomain der membrangebundenen Rezeptoren darstellen. Diese Rezeptoren sind dafür bekannt, häufig ihre extrazellulären Bindungsfunktionen zu behalten, wenn ihre Ektodomänen als rekombinantes löslichesProtein1 exprimiert werden. Für ein Protein von Interesse können lösliche rekombinante Proteine in jedem geeigneten eukaryotischen Proteinexpressionssystem in jedem Format hergestellt werden, vorausgesetzt, dass es für erhöhte Bindungsavidität oligomerisiert werden kann, und es enthält Tags, die in fluoreszenzbasierten Flusszytometrie-basierten Bindungstests verwendet werden können (z. B. FLAG-Tag, Biotin-Tag). Detaillierte Protokolle zur Herstellung von löslichen Ektodomains von Membranrezeptoren mit dem HEK293-Proteinexpressionssystem sowie verschiedene Multimerisierungstechniken und die Proteinexpressionskonstrukte für die Produktion sowohl pentamerischer Proteine als auch monomerer Proteine wurden zuvor beschrieben1,17. Das Protokoll beschreibt hier die Schritte zur Erzeugung fluoreszierender avider Sonden aus monomerer biotinylierter Proteine, indem sie zu Streptavidin konjugiert werden, das zu einem Fluorchrom konjugiert wird (z. B. Phycoerythrin oder PE), das direkt in zellbasierten Bindungstests verwendet werden kann und den Vorteil hat, dass kein sekundärer Antikörper zum Nachweis benötigt wird. Allgemeine Protokolle zur Durchführung genombezogener Screens wurden bereits20,21beschrieben, daher konzentriert sich das Protokoll hauptsächlich auf die Besonderheiten der Durchführung von Strömungszytometrie-basierten rekombinanten Proteinbindungssieben mit dem CRISPR/Cas9 Knockout Screening System mit der Human V1 ("Yusa") Bibliothek18.
Protokoll
1. Herstellung und Reinigung von biotinylierten His-tagged-Proteinen
- Verwenden Sie ein Proteinexpressionssystem auf Säugetier- oder Insektenzellbasis, um lösliche rekombinante biotinylierte Proteine zu produzieren (siehe Plasmidkonstrukte in Tabelle 1).
HINWEIS: Ein detailliertes Protokoll zur Herstellung von monomeren Biotin- und His-tagged-Proteinen mit dem HEK293-Zellexpressionssystem wird von Kerr et al.17beschrieben. Protein-Ektodomains, die mit dem HEK293-Expressionssystem exprimiert werden, werden in das Kulturmedium abgesondert. - Sammeln Sie die löslichen Proteine, indem Sie die Zellen 20 min durch Zentrifugieren bei 3.000 x g granulieren.
- Filtern Sie den Überstand durch einen 0,22-m-Filter und fügen Sie dem gefilterten Proteinüberstand die Ni2+-NTA Agaroseperlen im Verhältnis 1:1.000 (d. h. 50 l 50 % Agarose-Schlämme in 50 ml Überstand) hinzu. Über Nacht oder mindestens 4-5 h bei 4 °C auf einer rotierenden Plattform inkubieren.
- Waschen Sie die Polypropylensäule, indem Sie 5 ml His-Reinigungs-Waschpuffer hinzufügen. Siehe Tabelle 2 für alle Pufferzusammensetzungen.
- Gießen Sie die gesamte Perlenprotein-Überstandmischung in die Säule. Perlen sammeln sich an der Basis an.
- Waschen Sie die Perlen 2x mit 15 ml Waschpuffer. Um eine Proteinverdünnung zu vermeiden, ziehen Sie den Restwaschpuffer vorsichtig mit einer 5 ml Spritze aus der Säule und entsorgen Sie ihn.
- 300-500 L seines Reinigungs-Elutionspuffers vorsichtig direkt in die Perlen geben und mindestens 1 h brüten. Sammeln Sie das eluierte Protein, indem Sie die Flüssigkeit erneut sorgfältig mit einer 1 ml Spritze herausziehen. Tauschen Sie den Elutionspuffer mithilfe von Entsalzungsspalten in den gewünschten Puffer (z. B. normalerweise PBS oder HBS) aus. Alle Proteine bis zur weiteren Verwendung bei 4 °C lagern.
2. Quantifizierung und Oligomerisierung monomerer biotinylierter Proteine
ANMERKUNG: Um die Bindungs-Avidität zu erhöhen, oligomerisieren biotinylatierte monomere Proteine auf tetramerer Streptavidin-PE, bevor sie in Bindungstests verwendet werden. Erzielen Sie optimale Konjugationsverhältnisse von monomeren Proteinen und tetramerischem Streptavidin-PE, indem Sie eine Verdünnungsreihe von biotinylierten Monomeren gegen eine feste Konzentration von Streptavidin testen und empirisch die minimale Verdünnung ermitteln, bei der keine überschüssigen biotinylierten Monomere nachgewiesen werden können.
- Machen Sie mindestens acht serielle Verdünnungen von biotinylierten Proteinproben mit einem geeigneten Verdünnungspuffer (entweder PBS oder HBS mit 1% Rinderserumalbumin [BSA]) in einer 96-Well-Platte. Stellen Sie sicher, dass das Endvolumen jeder Verdünnung mindestens 200 l beträgt.
- Machen Sie eine doppelte Platte der Proben, indem Sie 100 l aus jedem Brunnen entfernen und in eine neue 96-Well-Platte übertragen. Fügen Sie immer ein Steuerelement ein. In diesem Fall sind Die Kontrollen reine Tag-Proteine (d. h. biotinylierte s.a. cd4-Domain 3+4-Protein). Dies wird als Kontrollsonde in allen Bindungstests verwendet.
- Verdünnen Sie Streptavidin-PE im Verdünnungspuffer auf 0,1 g/ml.
- Auf nur einer der Platten 100 l des verdünnten Streptavidin-PE hinzufügen. Die doppelte Platte dient als Steuerung. Fügen Sie 100 L Verdünnungspuffer in die Steuerplatte ein, um die Volumina auszugleichen.
- Inkubieren Sie für 20 min bei Raumtemperatur (RT). In der Zwischenzeit die Brunnen einer Streptavidin-beschichteten Platte mit dem Verdünnungspuffer für 15 min blockieren.
- Übertragen Sie das Gesamtvolumen der Probe von beiden Platten auf einzelne Brunnen der mit Streptavidin beschichteten Platten und inkubieren Sie für 1 h bei RT.
- Waschen Sie die Platte 3x mit 200 l Waschpuffer (d. h. entweder PBS oder HBS mit 0,1% Tween-20, 2% BSA). Fügen Sie 100 l von 2 g/mL Maus Anti-Ratten-Cd4d3+4 IgG (OX68) und inkubieren für 1 h bei RT.
- Waschen Sie die Platte 3x mit dem Waschpuffer. Fügen Sie 100 l eines Anti-Maus-Alkaliphosphatase-Konjugats bei 0,2 g/ml für 1 h bei RT hinzu.
- Waschen Sie die Platte 3x mit Waschpuffer und 1x im Verdünnungspuffer.
- Bereiten Sie p-Nitrophenylphosphat bei 1 mg/ml im Diethanolaminpuffer vor. Fügen Sie 100 l in jedem Brunnen hinzu und brüten Sie 15 min.
- Nehmen Sie Absorptionswerte bei 405 nm. Verwenden Sie die minimale Verdünnung, bei der kein Signal auf der Platte als geeigneter Verdünnungsfaktor vorhanden ist, um Tetramere zu erzeugen (Abbildung 2).
- Machen Sie eine 10-fache Tetramer-Färbungslösung für alle Proben und Kontrollen, indem Sie 4 g/ml Streptavidin-PE und die entsprechende biotinylierte Proteinverdünnung für 30 min bei RT inkubieren. Bewahren Sie konjugierte Proteine in einem lichtgeschützten Röhrchen bei 4 °C bis zur weiteren Verwendung auf.
3. Flow-Zytometrie-basierte Zellbindungs-Assays
- Für anhaftende Zellen, entfernen Sie Kulturmedien und waschen 1x mit PBS ohne divalente Kationen. Fügen Sie dann Zellablösungslösungen (z. B. EDTA) hinzu. Lassen Sie die Zellen für 5-10 min lösen. Tippen Sie vorsichtig auf den Kolben, um die Zellen freizugeben.
HINWEIS: Vermeiden Sie die Verwendung von Trypsin-basierten Produkten, da sie Zelloberflächenproteine spalten können. - Sammeln Sie getrennte Zellen in einem Rohr. Für Zellen, die in Suspension wachsen (z. B. HEK293-Zellen), sammeln Sie die Zellen direkt aus Kulturkolben in einem Rohr.
- Pelletzellen bei 200 x g für 5 min. Entfernen Sie den Überstand und setzen Sie das Pellet im Waschpuffer wieder auf (d.h. PBS/1% BSA).
- Zählen Sie die Zellen mit einem Hämozytometer und stellen Sie die Konzentration auf 2,5 x 105-1 x 106 Zellen/ml ein. Aliquot 100 l vorbereiteter Zellmischung auf einer 96 gut U- oder V-Bodenplatte. Drehen Sie die Platte für 5 min bei 400 x g. Entfernen Sie den Überstand mit einer Mehrkanalpipette.
- 100 l normalisierte fluoreszierend markierte hochavische Proteinsonden und Kontrollen in die zuvor vorbereiteten Platten mit Zellen geben und 1 h bei 4 °C inkubieren. Nach der Bindung für 1 h, drehen Sie die Platte bei 400 x g für 5 min.
- Entfernen Sie den Überstand, und fügen Sie 200 L Waschpuffer (d. h. PBS/1% BSA) hinzu. Gut mischen, indem Sie nach oben und unten pfeifen.
- Pellet die Zellen durch Zentrifugation bei 400 x g für 5 min. Wiederholen Sie den Waschschritt 1x. Nach zwei Washes den Überstand vollständig entfernen und das Zellpellet in 100 L PBS wieder aufhängen.
- Analysieren Sie die Zellen durch Durchflusszytometrie. Verwenden Sie den gelbgrünen Laser (d. h. 561 nm), um die PE-Fluoreszenz zu erkennen.
- Analysieren Sie zunächst die Zellen, die mit der Kontrollsonde befleckt wurden. Basierend auf der Verteilung der PE-Fluoreszenz, ziehen Sie ein Tor für die Bindung Spopulation so, dass nicht mehr als 1% der Kontrollzelle fällt in diesem Tor.
- Analysieren Sie die Probe, und bestimmen Sie den Zellanteil, der in das Bindungstor fällt.
HINWEIS: Zelllinien, die eine höhere Bindungspopulation anzeigen, sind für genetische Bildschirme erwünscht, da sie ein höheres Signal-Rausch-Verhältnis aufweisen. Idealerweise sollten über 80% der Zellen innerhalb dieses Tores liegen.
4. Bestimmung der Bindungsbeiträge von hitzelabilen Epitopen und Heparansulfat-Seitenketten
HINWEIS: Die Aktivität vieler Proteine ist hitzelabil, daher ist der Verlust der Bindungsaktivität nach der Wärmebehandlung ermutigend. Es wird empfohlen, den Beitrag von negativ geladenen Glykosaminoglykanen, hauptsächlich Heparansulfat (HS), bei der Vermittlung der Bindung der rekombinanten Proteine zu bestimmen. Dies liegt daran, dass die Bindung durch HS in dem hier beschriebenen Zellbindungstest additiv und nicht von anderen Rezeptoren koabhängig sein kann19. Dies bedeutet, dass die beobachtete Bindung vollständig durch HS-Seitenketten von Zelloberflächenproteoglykanen und nicht durch einen bestimmten Rezeptor vermittelt werden könnte. Die Bindung an HS auf der Zelloberfläche ist nicht unbedingt unspezifisch, sondern vielmehr eine Eigenschaft eines Proteins, die vor der Durchführung eines vollständigen genetischen Bildschirms zu wissen ist.
- Bereiten Sie wärmebehandelte Proteinproben für Bindungstests vor.
- Erhitzen Sie das normalisierte, aber nicht konjugierte monomere Protein bei 80 °C 10 min.
- Konjugieren Sie das wärmebehandelte Protein zu Streptavidin-PE unter der Annahme des gleichen Konjugationsverhältnisses wie sein unbehandeltes Gegenstück, wie durch ELISA bestimmt (siehe Abschnitt 2).
- Bereiten Sie Heparin-geblockte Proteinproben vor.
- Bereiten Sie acht 1:3 Verdünnungen von löslichem Heparin in PBS mit einer Anfangskonzentration von 2 mg/ml und einem Endvolumen von 100 l vor.
- Inkubieren Sie 100 l vorbereitete Bindungssonden in den Heparin-Verdünnungen für mindestens 30 min.
- Verwenden Sie in den in Abschnitt 3 beschriebenen Bindungstests wärmebehandeltes Protein und die vollen 200 l Heparin/Protein-Mischung. Repräsentative Ergebnisse sind in Abbildung 3A,Bdargestellt.
5. Auswahl von Zelllinien stabil ausdrückenCas9
HINWEIS: Bevor die Zelllinie, die die Sonde von Interesse bindet, in CRISPR-Screening verwendet werden kann, muss sie zuerst entwickelt werden, um die Cas9-Nuklease und einen hochaktiven Klon auszuwählen19auszudrücken.
- Verwenden Sie das folgende allgemeine Lentivirus-Produktionsprotokoll, um Lentivirus unter Verwendung des lentiviralen Konstrukts für die Cas9-Expression herzustellen (siehe Tabelle 1).
- Kultur HEK293-FT-Zellen in DMEM/10% FBS-Medien bei 37 °C und 5%CO2. Saat HEK293-FT-Zellen 1 Tag vor der Transfektion, so dass sie am Tag der Transfektion zu 80 % konfluent sind.
HINWEIS: HEK293FT-Zellen sind lose haftend; Wenn sie daher für die Herstellung von Lentiviren verwendet werden, sollten Sie daher erwägen, sie auf Kulturkolben zu beschichten, die mit 0,1% (w/v) Gelatine beschichtet sind, um die Haftung zu erhöhen. - Führen Sie Transfektionen am Morgen durch. Fügen Sie Transfervektor, Verpackungsmix und Transfektionsreagenz in vorgewärmte transfektionskompatible Medien (z. B. Opti-MEM) ein. Mischen Sie durch Invertieren der Röhre 10-15x. Inkubieren Sie für 5 min bei RT. Siehe Tabelle 3 für genaue Volumina.
- Fügen Sie das Transfektionsreagenz hinzu, wie vom Hersteller vorgeschlagen. Mix durch schnelles Wirbeln. 30 min bei RT inkubieren.
- Sehr sorgfältig aspirieren das verbrauchte Medium. Fügen Sie transfetionskompatible Medien zur Platte hinzu.
- Fügen Sie das Transfektionsreagenz/DNA-Komplexe tropfenweise auf die Seite der Platte und verbreiten Sie langsam durch die Platte durch wirbeln sehr sanft.
- Bei 37 °C für 3-5 h inkubieren und das Medium durch D10 medium ersetzen. Über Nacht inkubieren.
- Am nächsten Tag am Morgen, ersetzen Sie das Medium mit frischen D10 Medium. Über Nacht inkubieren.
- Am nächsten Tag spät am Nachmittag, sammeln Sie den viralen Überstand. Filter mit einem 0,45-mm-Filter mit geringer Proteinbindung. Optional frisches D10-Medium hinzufügen, über Nacht bebrüten und sich am nächsten Tag an den Überstand erinnern.
- Virusüberstand ist nur für wenige Tage bei 4 °C stabil. Bei -80 °C für langzeitbereite Lagerung lagern.
ANMERKUNG: Um ein hochkonzentriertes lentivirales Präparat zu erzeugen, das für die Transduktion von schwer zu transduzierenden Zellen wünschenswert sein könnte, können Überschärzeichen auch durch Zentrifugation bei 6.000 x g über Nacht bei 4 °C konzentriert werden. Markieren Sie das transluzente virushaltige Pellet mit einem Ethanol-resistenten Stift und entsorgen Sie den Überstand. Setzen Sie das Pellet in 1/100 des ursprünglichen Volumens für eine 100-fache Erhöhung der Konzentration aus.
- Kultur HEK293-FT-Zellen in DMEM/10% FBS-Medien bei 37 °C und 5%CO2. Saat HEK293-FT-Zellen 1 Tag vor der Transfektion, so dass sie am Tag der Transfektion zu 80 % konfluent sind.
- Transduce die Zellen mit Lentiviren.
- Platte 1 x 106 Zellen pro Brunnen in einer 6-Well-Platte mit 3 ml entsprechender Kulturmedien. Einige Zellen sind leichter transduziert als andere. Zur einfachen Transduce-Zellen (z.B. HEK-Zellen) fügen Sie das Lentivirus direkt zu den Zellen hinzu. Für schwierige Zellen, könnte es notwendig sein, ein Spinokulationsprotokoll zu folgen, wie unten beschrieben.
- Aliquot 2 ml von 2-5 x 106 Zellen/ml in einem 15 ml konischen Rohr.
- Fügen Sie das Lentivirus zusammen mit 8 g/ml Hexadimethrinbromid hinzu und inkubieren Sie bei RT für 30 min.
- Zentrifuge für 100 min bei 800 x g bei 32 °C. Anschließend wurden die Zellen im selben Medium wieder ausgesetzt und die Zellsuspension in entsprechende Kulturkolben mit entsprechenden Medien eingefügt.
- Transduktionen für mindestens 24 h zulassen. Anschließend entfernen Sie die Medien, die das Virus enthalten, und fügen Sie frisches Medium hinzu.
- Nach weiteren 24 h, ändern Sie die Medien zu einem, das mit den entsprechenden Antibiotika ergänzt wird. Das Cas9-Konstrukt enthält eine Blasticidin-Widerstandskassette zur Auswahl.
HINWEIS: Die Menge an Blasticidin muss für jede Zelllinie optimiert werden, indem eine Dosis-Wirkungs-Kill-Kurve ausgeführt wird. Eine Blasticidin-Konzentration zwischen 2,5-50 g/ml sollte die meisten untransduced Zelllinien innerhalb von 10 Tagen nach der Transduktion abtöten.
- Platte 1 x 106 Zellen pro Brunnen in einer 6-Well-Platte mit 3 ml entsprechender Kulturmedien. Einige Zellen sind leichter transduziert als andere. Zur einfachen Transduce-Zellen (z.B. HEK-Zellen) fügen Sie das Lentivirus direkt zu den Zellen hinzu. Für schwierige Zellen, könnte es notwendig sein, ein Spinokulationsprotokoll zu folgen, wie unten beschrieben.
- Durchführung der Selektion, bis alle Zellen in der Kontrollplatte (d. h. nicht transduzierte Zellen, die mit der gleichen Konzentration von Selektionsantibiotika behandelt wurden) abgetötet werden.
6. Auswahl von Klonen mit hoher Cas9-Aktivität
HINWEIS: Polyklonal Cas9 kann verwendet werden, um genetische Screens erfolgreich durchzuführen; Die Auswahl eines Klons mit hoher Cas9-Aktivität verbessert jedoch die Screening-Ergebnisse18.
- Verwenden Sie die Begrenzung der Verdünnung oder einzellige Sortierung einzelner blasticidinresistenter Zellen in Brunnen von drei 96 Brunnenplatten, die Kulturmedien enthalten, die mit Blasticidin ergänzt werden. Klone werden zwischen 2-4 Wochen entstehen. Wählen Sie 10-20 Klone und erweitern Sie in 6 Well-Platten.
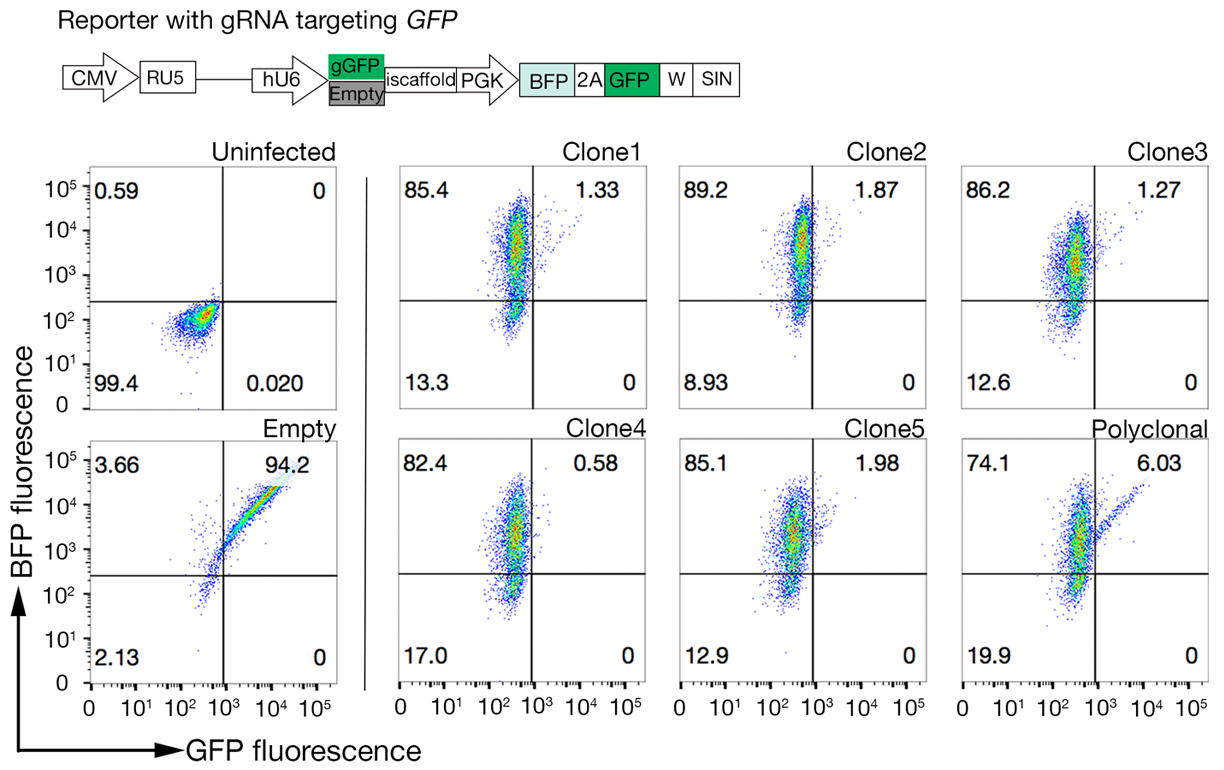
- Testen Sie die Klone für die Cas9-Aktivität mit dem schnell zu bewertenden GFP-BFP-System (green fluorescent protein-blue fluorescent protein), das ein exogenes Gen-Knockout-System verwendet, bei dem Zellen entweder mit einem Konstrukt, das GFP mit einem gRNA-targeting GFP exemittiert, oder einer leeren gRNA als Kontrollmethode18transduziert werden.
- Reporterplasmide bestellen: GFP-BFP-Plasmid, Control-BFP-Plasmid (Tabelle 1).
- Produzieren Sie Lentivirus sowohl für das GFP-BFP-Plasmid als auch für das Control-BFP-Plasmid unter Verwendung des in Abschnitt 5.1 beschriebenen Lentivirus-Produktionsprotokolls.
- Transduce jeden Cas9-exezierenden Zelllinienklon mit dem Lentivirus, das das GFP-BFP-System und Control-BFP separat kodiert. Folgen Sie dem Protokoll in Abschnitt 5.2.
- Untersuchen Sie nach 3 Tagen Transduktion die GFP-BFP-Fluoreszenz jedes Klons mittels Durchflusszytometrie. Verwenden Sie 488 nm Laser und 405 nm Laser, um GFP bzw. BFP zu erkennen.
- Quantitieren Sie die Cas9-Aktivität in jedem Klon, indem Sie das Verhältnis von BFP nur zu GFP-BFP-double-positiven Zellen untersuchen. Großaktivität Cas9-Zellen sollten idealerweise >95% GFP-Knockout-Effizienz haben (Abbildung 4).
7. Generierung genomweiter CRISPR-Cas9 Screening Knockout Bibliothek
- Für das genomweite Screening mit der Human V1-Bibliothek18bestellen Sie die genomweite Bibliothek (siehe Tabelle 1) und bereiten Sie die Plasmidbibliothek aus dem bakteriellen Stich unter Verwendung des Protokolls vor, das unter "Protokolle für bibliotheksreplikation" im Herstellerhandbuch bereitgestellt wird.
- Verwenden Sie die genomweite Bibliotheksplasmidpräparation, um eine lentivirale Bibliothek zu erstellen, die gRNAs für eine gezielte Störung menschlicher Gene mit dem in Abschnitt 5.1 beschriebenen Lentivirus-Produktionsprotokoll kodiert.
HINWEIS: Eine gute Praxis besteht darin, eine einzige Charge lentiviraler Präparation zu produzieren, die für die Transduktion optimiert ist, um die experimentelle Konsistenz zu verbessern. - Verwenden Sie das Transduktionsprotokoll in Abschnitt 5.2, um kleine Testtransduktionen durchzuführen, um die erforderliche Virusmenge für jede Zelllinie zu bestimmen, um eine Transduktion von 30 % zu erreichen. Verwenden Sie die Durchflusszytometrie, um die BFP-Fluoreszenz als Proxy für die Transduktionseffizienz zu bewerten.
- Um HEK293-Zellen zu transducen, fügen Sie einfach die vorbestimmte lentivirale Präparation zu 30-50 x 106 Zellen hinzu, die in normalen Wachstumsmedien für 4 h kultiviert werden. Dann entfernen Sie die Medien mit Lentivirus und ersetzen Sie durch frische Wachstumsmedien.
- Verwenden Sie für andere Zelllinien das Spinokulationsprotokoll in Abschnitt 5.2.1, jedoch in größerem Maßstab, sodass insgesamt 30-50 x 106 Zellen transduziert werden. Dazu aliquot 2 ml von 5 x 106 Zellen/ml in einem 15 ml konischen Rohr und gehen Wie angegeben vor.
- Wählen Sie für anhaftende Zelllinien transduzierte Zellen aus, indem Sie Puromycin 24 h nach der Transduktion hinzufügen.
HINWEIS: Optimieren Sie die Puromycin-Konzentrationen, indem Sie eine Dosis-Wirkungs-Kill-Kurve durchführen. Normalerweise sollten Konzentrationen zwischen 1-10 g/ml nicht transduzierte Zellen innerhalb von 3-5 Tagen abtöten. Vermeiden Sie höhere Konzentrationen von Puromycin, da dies die Chancen erhöhen kann, Zellen auszuwählen, die von mehr als einer einzigen Führungs-RNA (sgRNA) transduziert wurden. - Für Suspensionszellen, Ernte transduzierte (d.h. BFP-positive) Zellen 3 Tage posttransduktion mit einem Zellsortierer und generieren Bibliotheken, die mindestens 10 x 106 Zellen enthalten. Einmal mit BFP ausgewählt, wachsen die Zellen in Medien mit einer angemessenen Menge an Puromycin ergänzt.
HINWEIS: Vermeiden Sie Auswahlen nur mit Puromycin für Suspensionszelllinien, da es schwierig ist, abgestorbene Zellen und Ablagerungen aus Suspensionszellkulturen zu entfernen, die die Zellsortierung stören können. - Kultur mutierte Bibliothek für 9-16 Tage Posttransduktion mit regelmäßiger Passage alle 2-3 Tage.
8. Genetisches Screening zur Zelloberflächenbindung
- Pellet die mutierte Zellbibliothek bei 200 x g für 5 min und setzen Sie die Zellen in PBS wieder auf.
- Teilen Sie die Zellen in zwei 15 ml konische Röhren mit mindestens 50 x 106 Zellen in jeder Röhre.
- Drehen Sie ein konisches Rohr bei 200 x g für 5 min, entfernen Sie den Überstand und frieren Sie das Zellpellet bei -20 °C ein. Dies ist die Kontrollpopulation und wird später verarbeitet.
- Das Pellet in der anderen Röhre in 10 ml PBS/1% BSA wieder aufsetzen. Legen Sie 100 l Zellen als Negativkontrolle auf einer 96-Well-Platte beiseite.
- Fügen Sie das entsprechende vorkonjugierte rekombinante Protein der Zellsuspension in der konischen Röhre und negative Kontrollproteine in die 96-Well-Platte ein.
- Zellfärbung für mindestens 1 h bei 4 °C auf einem Tischrotor mit sanfter Drehung (6 Umdrehung) durchführen.
- Pellet die Zellen bei 200 x g für 5 min, entfernen Sie den Überstand. Führen Sie zwei Waschschritte aus, und setzen Sie die Zellen in 5 ml PBS erneut aus.
- Dehnen Sie die Zellen mit einem 30-m-Zellsieb, um Zellcluster zu entfernen. Analysieren Sie mit einem Flusssortierer.
- Verwenden Sie die negative Kontrollprobe zum Gate für BFP+/PE-Zellen.
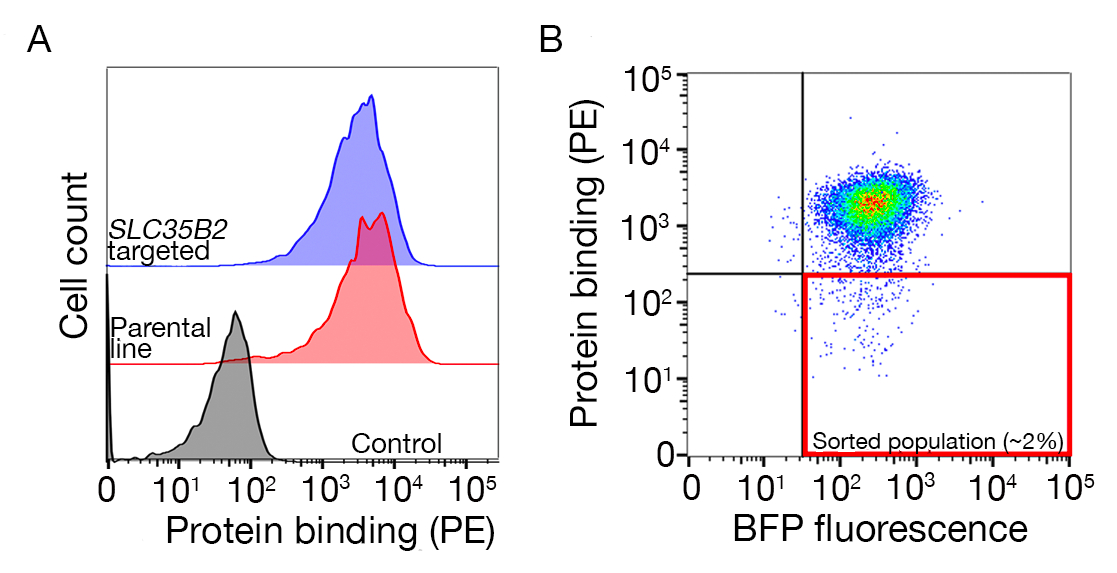
- Sortieren Sie die Probe und sammeln Sie die BFP+/PE-Zellen. Die Sortierungstore hängen von der Bindung der Zellen an das Protein ab, betragen aber normalerweise 1-5% der PE-negativen Proben. Ein Beispiel für ein Sortiertor finden Sie in Der Zusätzlichen Abbildung 1.
- Sammeln Sie 500.000-1.000.000 Zellen aus dem ausgewählten Tor. Angesichts der geringen Anzahl von Zellen sollten Sie die Proben in einem 1,5 ml Zentrifugationsrohr sammeln, um Verluste zu minimieren.
- Pellet die sortierten Zellen durch Zentrifugieren bei 500 x g für 5 min. Vorsichtig entfernen Sie den Überstand und entsorgen. Es ist möglich, das Pellet bis zu 6 Monate bei -20 °C zu lagern.
9. Genomische DNA-Extraktion und erste PCR zur gRNA-Anreicherung
- Extrahieren Sie genomische DNA aus der population mit hoher Komplexität.
- Wenn die Kontrollpopulation bei -20 °C eingefroren war, nehmen Sie das konische Rohr heraus und fügen Sie PBS hinzu. Auf Eis halten, um das Pellet aufzutauen.
- Verwenden Sie ein kommerzielles Kit (siehe Tabelle der Materialien) mit den Empfehlungen des Herstellers, um genomische DNA aus 50 x 106 Zellen zu extrahieren. Stellen Sie die DNA-Konzentration auf 1 mg/ml ein.
- Richten Sie für jede Probe einen Master-Mix für PCR ein, der 72 g DNA entspricht. Aliquot 50 l pro Brunnen in 36 Brunnen einer 96 Well PCR Platte. Die erforderlichen Primersequenzen sind in Tabelle 4aufgeführt. Verwenden Sie die Anleitung in Tabelle 5 und 6.
- Lösen Sie 5 l der PCR von 6-12 repräsentativen Proben auf einem 2% (w/v) Agaro-Gel auf. Es sollte ein einziges klares Band bei 250 bp beobachtet werden. Wenn die Bänder schwach sind, wiederholen Sie die PCR für weitere 2-3 Zyklen.
- Verwenden Sie eine Mehrkanalpipette, um 5 l PCR-Produkte aus jedem Bohrgut zu sammeln (insgesamt 180 L) und sie in einem Reservoir mit 900 l Bindungspuffer aus einem kommerziellen Kit zu bündeln (siehe Tabelle der Materialien).
- Reinigen Sie die PCR-Produkte mit einem kommerziellen PCR-Reinigungskit. Elute DNA in 50 l Elutionspuffer aus einem kommerziellen Kit (siehe Tabelle der Materialien) und messen Sie die DNA-Konzentration.
- Proben, die für den Verlust des Bindungsphänotyps sortiert wurden, bestehen wahrscheinlich nicht aus einer großen Anzahl unabhängiger Klone. Daher ist es nicht notwendig, PCR mit 72 g DNA durchzuführen. Isolieren Sie die DNA mit einem geeigneten kommerziellen Kit (siehe Tabelle der Materialien). Richten Sie 3-4 PCR-Reaktionen mit dem zuvor beschriebenen Protokoll (Abschnitt 9.1.3) mit 100 ng/l DNA ein. Wenn die sortierte Zellzahl weniger als 100.000 ist, verwenden Sie Zelllysate anstelle von genomischen DNA-Präparaten.
- Aliquot etwa 10.000 Zellen/gut in einer 96 well PCR-Platte.
- Pellet die Zellen in der Platte und sorgfältig entfernen die meisten der Überstand. Das Pellet ist nicht sichtbar.
- Fügen Sie in jedem Brunnen 25 l Wasser hinzu und erhitzen Sie die Proben bei 95 °C für 10 min.
- Fügen Sie jedem Brunnen für 1 h 5 l von 2 mg/ml frisch verdünnte Proteinase K hinzu und bei 56 °C zu brüten. Dann erhitzen Sie die Probe für 10 min bei 95 °C, um die Proteinase K zu inaktivieren.
- Verwenden Sie 10 l Zelllysatgemisch pro PCR-Reaktion. Lysate sollten innerhalb von 24 h verwendet werden.
10. Zweite PCR-Runde für Index-Barcoding und -Sequenzierung
- Verdünnen Sie die Produkte aus der ersten Runde PCR auf 40 pg/ l.
- Richten Sie eine PCR pro Probe ein (verwenden Sie die Anleitung in den Tabellen 7 und 8). Die Verwendung einer Hochtreuepolymerase ist wichtig, um Fehler zu minimieren, die durch die Polymerase während der sgRNA-Amplifikation auftreten.
- Lösen Sie 5 l PCR-Produkte auf einem 2% (w/v) Agaro-Gel auf. Ein einzelnes klares Band bei 330 bps sollte beobachtet werden.
- Reinigen Sie PCR-Produkte mit paramagnetischen Perlen, indem Sie den PCR-Produkten 31,5 l (0,7x Gesamtvolumen) resuspendierter Perlen hinzufügen, gut mischen und 5 min bei RT brüten.
- Legen Sie das Rohr für 3 min auf ein magnetisches Rack. Die Perlen sollten auf der Seite der Platte eingefangen werden und die Lösung sollte klar sein. Entfernen und entsorgen Sie den Überstand vorsichtig.
- 150 l mit 80 % frisch zubereitetem Ethanol in die Röhre geben. 30 s inkubieren und dann vorsichtig entfernen und entsorgen.
- Wiederholen Sie Schritt 13.6, diesmal mit 180 l. Dann trocknen Sie die Perlen 5 min an der Luft.
- Entfernen Sie das Rohr vom Magneten. Elute DNA-Ziel von Perlen in 35 L sterilen EB-Puffer. Inkubieren Sie für 3 min, dann legen Sie das Rohr wieder in das magnetische Rack für 3 min.
- Übertragen Sie ca. 30 l des Überstandes, der die eluierten PCR-Produkte enthält, in ein Röhrchen.
- Sequenzieren Sie die Beispiele auf einer Sequenzierungsplattform der nächsten Generation. Verwenden Sie für die HumanV1 gRNA-Bibliothek den in Tabelle 4 aufgeführten benutzerdefinierten Primer, um 19 bp zu sequenzieren.
11. Bioinformatik-Analyse zur Identifizierung des Rezeptors und der damit verbundenen Wege
- Ordnen Sie Sequenzen von einer sortierten und unsortierten Grundgesamtheit der Referenzbibliothek mithilfe der Zählfunktion von MAGeCK zu. Die Funktion liefert eine Rohzähldatei (Ergänzende Tabelle 1).
HINWEIS: Detaillierte Anweisungen zur Installation von MAGeCK und zur Nutzung verschiedener Funktionen innerhalb von MAGeCK sind in einem zuvor veröffentlichten Protokoll von Wang et al.20beschrieben. - Überprüfen Sie den technischen Standard der im Bildschirm verwendeten Steuerungsbibliothek.
- Median normalisieren die Rohanzahl und verwenden das ggplot2-Paket in R21 oder einer gleichwertigen Software, um ein empirisches kumulatives Dichtefunktionsdiagramm der Zählungen in Plasmid zu zeichnen und unsortierte Proben zu kontrollieren (Abbildung 5A).
- Führen Sie die -test-Funktion von MAGeCK aus, indem Sie Zählungen aus der Plasmidpopulation als "Kontrolle" und die Zählungen aus unsortierten Kontrollproben als "Test"-Probe verwenden. Die Funktion ergibt eine Genzusammenfassungsdatei (Ergänzende Tabelle 1).
- Öffnen Sie die Genzusammenfassungsdatei und zeichnen Sie die Verteilung von Log-Fold-Changes (neg|lfc-Spalte) für zuvor kategorisierte wesentliche und nicht-essentielle Gene22 (Abbildung 5B).
- Wählen Sie deutlich erschöpfte Gene (neg|fdr < 0.05) und führen Sie eine Signalanalyse mit dem Enrichr23-Paket oder gleichwertigen Pfadanreicherungspaketen in R durch (Abbildung 5C).
- Führen Sie die -test-Funktion von MAGeCK mit Standardeinstellung aus. Verwenden Sie Rohzählungen aus einer unsortierten Kontrollstichprobe als "Steuerelement" und Zählungen aus sortierter Probe als "Behandlung" bei der Durchführung der Analyse.
- Öffnen Sie die von MAGeCK generierte Genzusammenfassungsdatei, und ordnen Sie die Spalte pos|rank in aufsteigender Reihenfolge ein. Verwenden Sie FDR (pos|fdr-Spalte) < 0.05 als Cutoff zur Identifizierung von Treffern. Der Rezeptor wird in der Regel hoch eingestuft, oft in der ersten Position.
- Plotten Sie die Robust-Ranking-Algorithmus (RRA) Punkte für positive Auswahl (pos|score) in R oder einer gleichwertigen Software (Abbildung 5D).
- Wählen Sie die Gentreffer aus und führen Sie eine Signalanalyse mit dem Anreicherungspaket oder gleichwertigen Signalweganreicherungspaketen in R durch, um die angereicherten Pfade zu identifizieren.
Ergebnisse
Sequenzierungsdaten von zwei repräsentativen Genom-K.o.-Bildschirmen zur Identifizierung des Bindungspartners des menschlichen TNFSF9 und P. falciparum RH5, die in NCI-SNU-1 bzw. HEK293-Zellen durchgeführt werden, sind vorgesehen (Ergänzende Tabelle 1). Das Bindungsverhalten von RH5 wurde sowohl durch Heparansulfat als auch durch seinen bekannten Rezeptor BSG24 (Abbildung 3C) beeinflusst, während TNFRSF9 speziell an seinen bekannten Rezeptor TNFSF9 gebunden war und bei der Vorinkubation mit löslichem Heparin nicht die Bindung verlor. Protein 3 in Abbildung 3B steht für TNFRSF9.
Für beide Zelllinien ist auch die Verteilung von gRNAs in der Kontrollmutantenbibliothek nach 3 Tagen (9, 14 und 16 Tage Nachtransduktion) vorgesehen (Ergänzende Tabelle 1). Die gRNA-Verteilung zeigte, dass die Bibliothekskomplexität im Laufe des Experiments beibehalten wurde (Abbildung 5A). Der genetische Screen zur Identifizierung des Liganden für TNFSF9 wurde am Tag 14 der Posttransduktion durchgeführt, während für RH5 Tag 9 Posttransduktion durchgeführt wurde. Die technische Qualität der Screens wurde bewertet, indem die Verteilung der beobachteten Faltenveränderungen von gRNAs untersucht wurde, die auf einen Referenzsatz nicht wesentlicher Gene im Vergleich zur Verteilung für Referenzsatz wesentlicher Gene abzielen22 (Abbildung 5B). Darüber hinaus zeigte die Anreicherung auf Signalwegebene auch, dass erwartete wesentliche Pfade in der "Drop-out"-Population identifiziert und signifikant bereichert wurden, wenn man die Kontrollprobe mit der ursprünglichen Plasmidbibliothek vergleicht. Ein Beispiel mit dem Beispiel "Tag 14 NCI-SNU-1" ist in Abbildung 5Cdargestellt.
Die Verteilung der gRNAs in der kontrollierbaren versus sortierten Population mit der -test-Funktion von MAGeCK (siehe Ergänzende Tabelle 1 für die Genzusammenfassungsausgabe von MAGeCK) wurde verwendet, um den Rezeptor aus den phänotypischen Bildschirmen zu identifizieren. Der modifizierte RRA-Score, der von MAGeCK in der Gen-Level-Analyse gemeldet wird, wird anhand der Gene dargestellt, die nach p-Werten geordnet sind. Der RRA-Score in MAGeCK bietet eine Messgröße, bei der gRNAs konstant höher eingestuft werden als erwartet. Im Bildschirm für TNFRSF9 war tNFSF9 der Top-Hit, ein bekannter Bindepartner von TNFRSF9 (Abbildung 5D). Darüber hinaus wurden eine Reihe von Genen im Zusammenhang mit dem TP53-Signalweg identifiziert. Im Falle von RH5 wurde neben dem bekannten Rezeptor (BSG) und dem für die Herstellung der sulfatierten GAGs (SLC35B2) erforderlichen Gen auch ein zusätzliches Gen (SLC16A1) identifiziert (Abbildung 5E). SLC16A1 ist ein Chaperon, das für den Handel mit BSG an die Oberfläche von Zellen25benötigt wird. Zusammen zeigen diese Ergebnisse die Fähigkeit des Bildschirms, direkt interagierende Rezeptoren und die zellulären Komponenten zu identifizieren, die erforderlich sind, damit dieser Rezeptor auf der Oberfläche der Zellen in funktioneller Form exprimiert wird.

Abbildung 1: Überblick über den genetischen Screening-Ansatz zur Identifizierung von Zelloberflächenrezeptoren. Dieser Test besteht aus drei Hauptschritten: Erstens werden rekombinante Proteine, die die Ektodomäne von Zelloberflächenrezeptoren darstellen, in einer Zelllinie exprimiert, die strukturkritische posttranslationale Modifikationen wie HEK293-Zellen hinzufügen kann. Monomere Protein-Ektodomains werden oligomerisiert, indem sie zu Streptavidin-PE konjugieren, um ihre Bindungs-Avidität zu erhöhen. Zweitens werden diese avianen Sonden in zellulären Bindungstests verwendet, bei denen helle Färbungen an den Zelllinien, die durch eine prominente Verschiebung der PE-Fluoreszenz (in Grün) im Vergleich zu einem Negativkontrollprotein (in Schwarz) angezeigt werden, das Vorhandensein eines Zelloberflächenbindungspartners zeigen. Drittens werden rezeptorpositive Cas9-exemittierende Zelllinien ausgewählt und ein Genom-Skalen-Screening mit gRNAs durchgeführt, die auf die überwiegende Mehrheit der proteinkodierenden Gene abzielen. Bei der Erzeugung von mutierten Bibliotheken ist es üblich, 30% Transduktionseffizienz zu verwenden, die auf der Poisson-Verteilungswahrscheinlichkeit basiert, die sicherstellt, dass jede Zelle eine einzelne gRNA erhält, so dass der resultierende Phänotyp einem bestimmten Knockout zugeschrieben wird. Der von den transduzierten Zellen ausgedrückte BFP-Marker wird verwendet, um Zellen auszuwählen, die gRNAs mit FACS enthalten. Phänotypic Bildschirme werden zwischen 9-16 Tage posttransduktion durchgeführt. Am Tag des Bildschirms wird die gesamte mutierte Zellpopulation in zwei geteilt. Die eine Hälfte wird als Kontrollpopulation gehalten und die andere Hälfte wird für die rekombinante Proteinbindung ausgewählt. Die Zellen aus der mutierten Bibliothek, die das rekombinante Protein nicht mehr binden können, werden mit FACS sortiert, und die Anreicherung von gRNAs in der sortierten versus Kontrollpopulation wird verwendet, um Gene zu identifizieren, die für die Zelloberflächenbindung der beschrifteten Avi-Sonde erforderlich sind. Schritte im Protokoll, die viel Zeit in Anspruch erfordern, werden angezeigt. Diese Zahl wurde von Sharma et al.19geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Ermittlung des Verhältnisses von biotinyliertem Protein zu Streptavidin-PE mit einer ELISA-basierten Methode. Ein Beispiel für die Konjugationsstrategie von Streptavidin-PE, mit der eine aviante Sonde aus einem biotinylierten monomeren Protein erzeugt wird. Eine Verdünnungsserie von biotinylierten Monomeren wurde gegen eine feste Konzentration von Streptavidin inkubiert. Die minimale Verwässerung, bei der keine überschüssigen biotinylierten Monomere nachgewiesen werden können, wurde durch ELISA ermittelt. ELISA wurde mit oder ohne Vorinkubation einer Reihe von Proteinverdünnungen mit 10 ng Streptavidin-PE durchgeführt. In Gegenwart von Streptavidin-PE wurde die minimale Verdünnung, bei der kein Signal identifiziert wurde (umkreist schwarz) und die für die Sättigung erforderliche Proteinmenge berechnet, um eine 10-fache Stammlösung mit 4 g/ml Streptavidin-PE zu erzeugen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Repräsentative Bindung von Proteinen an Zelllinien. (A) Die Proteinbindung an Zelllinien hatte im Vergleich zur Kontrollprobe einen deutlichen Anstieg der zellassoziierten Fluoreszenz. Die Wärmebehandlung (80 °C für 10 min) des rekombinanten Proteins brach alle Bindungen an eine negative Kontrolle ab, was zeigt, dass das Bindungsverhalten von richtig gefaltetem Protein abhängig war. (B) Verschiedene Klassen des Proteinbindungsverhaltens an Zelloberflächen; Abhängigkeit von GAGs. Von links nach rechts können die Proteine in drei Typen eingeteilt werden: Protein Typ 1 adsorbiert nur an HS. Diese Proteine verlieren ihre Bindung nach der Präinkubation mit Heparinkonzentrationen über 0,2 mg/ml. Protein Typ 2 bindet zusätzlich zu einem bestimmten Rezeptor an HS. Diese Proteine verlieren in den Preblockierungsexperimenten teilweise an Bindung. Protein Typ 3 bindet HS nicht. Diese Proteine verlieren nicht bindungsfrei im Vergleich zu Elternlinien. (C) Ein Beispiel für ein Protein (d. h. RH5), das auf additiv an HS und einen spezifischen Rezeptor bindet. Die Ausrichtung entweder auf den Rezeptor (d. h. BSG) oder auf Enzyme, die für die HS-Synthese erforderlich sind (z. B. SLC35B2, EXTL3), reduziert die Bindung von RH5 an Zellen im Verhältnis zu Kontrollen nur teilweise. Transduzierte polyklonale Linien können in solchen Experimenten verwendet werden, um ein Bindungsverhalten zu etablieren. Diese Zahl wurde von Sharma et al.19geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Auswählen von klonalen Zelllinien mit hoher Cas9-Aktivität. Die Genom-Editing-Effizienz sowohl von polyklonalen als auch von geklonten Linien von NCI-SNU-1-Zelllinien wurde mit dem GFP-BFP-Reportersystem bewertet, bei dem Zelllinien mit Viren mit einem gRNA-zielgerichteten Plasmid kodiert GFP oder ohne (d. h. "leer") transduziert wurden. Ein Schaltplan wird dargestellt. Die Durchflusszytometrie wurde verwendet, um sowohl die BFP- als auch die GFP-Expression nach der Transduktion zu testen und mit der nicht infizierten Kontrolle verglichen zu werden. Der GFP-Ausdruck wurde als Proxy für die Cas9-Aktivität verwendet, während BFP-Expression als transduzierte Zellen markiert wurde. Das Profil für nicht infizierte und leere infizierte Zellen sah für alle Klone ähnlich aus. Repräsentative Profile sind im linken Bereich dargestellt. Alle fünf Klone der NCI-SNU-1-Zelllinie wiesen einen höheren GFP-Verlust im Vergleich zur polyklonalen Linie (rechtes Panel) auf, wobei Klon 4 die höchste Effizienz bei der niedrigsten feuerfesten Population zeigte. Diese Zahl wurde von Sharma et al.19geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Repräsentative Ergebnisse aus genetischen Screens zur Identifizierung der Zelloberflächenbindungspartner. (A) Kumulative Verteilungsfunktionsdiagramme, die die gRNA-Fülle in der Plasmidbibliothek mit den mutierten Bibliotheken der HEK-293-E- und NCI-SNU-1-Zellen am Tag 9, 14 und 16 Tagen nach der Transduktion vergleichen. Für jede bestimmte Zahl meldet die funktion der kumulativen Dichte den Prozentsatz der Datenpunkte, die unter diesem Schwellenwert lagen. Die kleine Verschiebung der mutierten Zellpopulation im Vergleich zur ursprünglichen Plasmidpopulation stellt die Erschöpfung in einer Teilmenge von gRNAs im Vergleich zur Plasmidbibliothek dar. (B) Verteilung von Log-Fold-Änderungen in Genen, die zuvor in den Zelllinien HEK293 und NCI-SNU-1 als wesentlich (rot) oder nicht wesentlich (schwarz) kategorisiert wurden. Die Verteilung der Faltenwechsel für nicht essentielle Gene zentrierte sich auf 0 , während sich die Verteilung für wesentliche Gene nach links in Richtung negativer Faltenveränderungen verlagerte. (C) Signifikant angereicherte Pfade in Genen, die in der NCI-SNU-1-Mutantenkontrollpopulation 14 Tage nach der Transduktion erschöpft sind. Erwartete bekannte zellessentielle Pfade wurden identifiziert. (D) Robust Rank Algorithm (RRA)-Score für Gene, die in den sortierten Zellen angereichert wurden, die die Fähigkeit verloren hatten, die TNFRSF9-Sonde zu binden. Gene wurden nach dem RRA-Score gewertet. Der bekannte Interaktionspartner TNFSF9 und Gene im Zusammenhang mit dem TP53-Signalweg (rot markiert) wurden im Bildschirm identifiziert. (E) Ranggeordnete RRA-Scores für Gene, die aus der gRNA-Anreicherungsanalyse identifiziert wurden, die für die RH5-Bindung an HEK293-Zellen erforderlich sind (linkes Panel). SLC35B2 und SLC16A1 wurden innerhalb eines FDR-Schwellenwerts (False-Discovery-Rate) von 5 % identifiziert. Zwei weitere Gene im HS-Biosyntheseweg (z. B. EXTL3 und NDST1) wurden innerhalb der FDR von 25% identifiziert. Schematisch darstellung den allgemeinen GAG-Biosyntheseweg mit den entsprechenden Genen, die den entsprechenden Schritten zugeordnet sind (Panel 2). Gene, die für die Verpflichtung zur Chondroitinsulfat-Biogenese (d. h. CSGALNACT1/2) erforderlich sind, wurden im Bildschirm nicht identifiziert. Diese Zahl wurde von Sharma et al.19geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Plasmidname | Plasmid # | Verwenden |
| Proteinexpressionskonstrukt: CD200RCD4d3+4-bio-linker-his | Addgen: 36153 | Herstellung von rekombinantem Protein mit CD4d3+4, Biotin und 6-his-Tags. |
| pMD2.G | Addgen: 12259 | VSV-G-Hüllkurve, die Plasmid exzessiumdrückt; Produktion von Lentivirus |
| psPAX2 | Addgen: 12260 | Lentivirales Verpackungsplasmid, Herstellung von Lentivirus |
| Cas9-Konstruktion: pKLV2-EF1a-Cas9Bsd-W | Addgen: 68343 | Produktion der konstitutiv ausdrückenden Cas9-Linie |
| gRNA-Expressionskonstrukt: pKLV2-U6gRNA5(BbsI)-PGKpuro2ABFP-W | Addgen: 67974 | CRISPR gRNA Expressionsvektor mit verbessertem Gerüst und Puro/BFP-Markern |
| Human Improved Genome-wide Knockout CRISPR Library | Addgen: 67989 | Eine gRNA-Bibliothek gegen 18.010 menschliche Gene, die für den Einsatz im Lentivirus entwickelt wurden. |
| GFP-BFP-Konstrukt: pKLV2-U6gRNA5(gGFP)-PGKBFP2AGFP-W | Addgen: 67980 | Cas9-Aktivitätsreporter bei BFP und GFP. |
| Leerbau: pKLV2-U6gRNA5(empty)-PGKBFP2AGFP-W | Addgen: 67979 | Cas9-Aktivitätsreporter (Kontrolle) mit BFP und GFP. |
Tabelle 1: Plasmide, die bei diesem Ansatz verwendet werden.
| Puffername | Komponenten |
| HBS (10X) | 1,5 M NaCl und 200 mM HEPES in MiliQ Wasser, auf pH 7,4 einstellen |
| PBS (10X) | 80 g NaCl, 2 g KCl, 14,4 g Na2HPO4 und 2,4 g KH2PO4 in MiliQ Wasser, pH 7,4 |
| Natriumphosphatpuffer (80mM Lager) | 7.1 g Na2HPO4.2H2O, 5.55 g NaH2PO4, pH 7.4 |
| Sein Reinigungsbindungspuffer | 20 mM Natriumphosphatpuffer, 0,5 M NaCl und 20 mM Imidazol, pH 7,4 |
| Sein Reinigungs-Elutionspuffer | 20 mM Natriumphosphatpuffer, 0,5M NaCl und 400 mM Imidazol, pH 7,4 |
| Diethanolamin-Puffer | 10% Sthanamamin und 0,5 mM MgCl2 in MiliQ-Wasser, auf pH 9,2 einstellen: |
| D10 | DMEM, 1% Penicillin-Streptomycin (100 Einheiten/ml) und 10% hitzeinaktiviertes FBS |
Tabelle 2: Puffer, die für diese Studie erforderlich sind.
| Komponenten | 10-cm-Schale | 6-Well-Platte |
| 293FT Zellen | 70–80% konfluent | 70–80% konfluent |
| Transfetionskompatible Medien (Opti-MEM) (Schritt 5.1.2) | 3 mL | 500 l |
| Transfetionskompatible Medien (Opti-MEM) (Schritt 5.1.4) | 5 mL | 2 mL |
| Lentiviraler Transfervektor | 3 g | 0,5 g |
| psPax2 (siehe Tabelle 1) | 7,4 g | 1,2 g |
| pMD2.G (siehe Tabelle 1) | 1,6 g | 0,25 g |
| PLUS-Reagenz | 12 l | 2 l |
| Lipofectamin LTX | 36 l | 6 l |
| D10 (Schritt 7.1.7) | 5 mL | 1,5 ml |
| D10 (Schritt 7.1.8 und 7.1.10) | 8 mL | 2 mL |
Tabelle 3: Mengen und Mengen von Reagenzien für den Lentivirus-Verpackungsmix.
Tabelle 4: Primersequenzen zur Verstärkung von gRNA und NGS. Bitte klicken Sie hier, um diese Datei anzuzeigen (Rechtsklick zum Herunterladen).
| Reagenz | Volumen pro Reaktion | Master-Mix (x38) |
| Q5 Hot Start High-Fidelity 2x | 25 l | 950 l |
| Primer (L1/U1) Mischung (je 10 m) | 1 L | 38 l |
| Genomische DNA (1 mg/ml) | 2 l | 72 l |
| H2O | 22 l | 1100 l |
| gesamt | 50 l | 1900 'L |
Tabelle 5: PCR zur Verstärkung von gRNAs aus Proben mit hoher Komplexität.
| Zyklusnummer | Denature | Glühen | Erweiterung |
| 1 | 98 °C, 30er Jahre | ||
| 2-24 | 98 °C, 10s | 61 °C, 15s | 72 °C, 20er Jahre |
| 25 | 72 °C, 2 min |
Tabelle 6: PCR-Bedingungen für die erste PCR.
| Reagenz | Volumen pro Reaktion |
| KAPA HiFi HotStart ReadyMix | 25 l |
| Primer (PE1.0/Indexprimer) Mix (je 5 m) | 2 L |
| Erstes PCR-Produkt (40 pg/l) | 5 l |
| H2O | 18 l |
| gesamt | 50 l |
Tabelle 7: PCR für die Index-Tagging von sgRNAs aus genetischen Bildschirmen.
| Zyklusnummer | Denature | Glühen | Erweiterung |
| 1 | 98 °C, 30er Jahre | ||
| 2-15 | 98 °C, 10s | 66 °C, 15s | 72 °C, 20er Jahre |
| 16 | 72 °C, 5 min |
Tabelle 8: PCR-Bedingungen für die zweite PCR.

Ergänzende Abbildung S1: Eine Anleitung zum Zeichnen von Toren zum Sortieren der unverbindlichen Population. (A) Ein idealer Proteinkandidat für das Screening sollte eine deutliche Verschiebung der Bindungspopulation im Vergleich zur Kontrollpopulation haben, und die Bindung sollte auf Zellen beibehalten werden, denen Maschinen für die HS-Biosynthese fehlen. Anstelle von Tests an SLC35B2-Zielzelllinien kann ein Heparin-Blockierungsexperiment verwendet werden. (B) Es wurden Zellen gesammelt, die keine Oberflächenfärbung des Proteins ectodomain, aber bFP-Fluoreszenz aus lentiviraler Transduktion ausdrücken. Die angezeigten Zellen sind von einem Bildschirm für die Identifizierung von Rezeptor für GABBR222. Diese Zahl wurde von Sharma et al.19geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

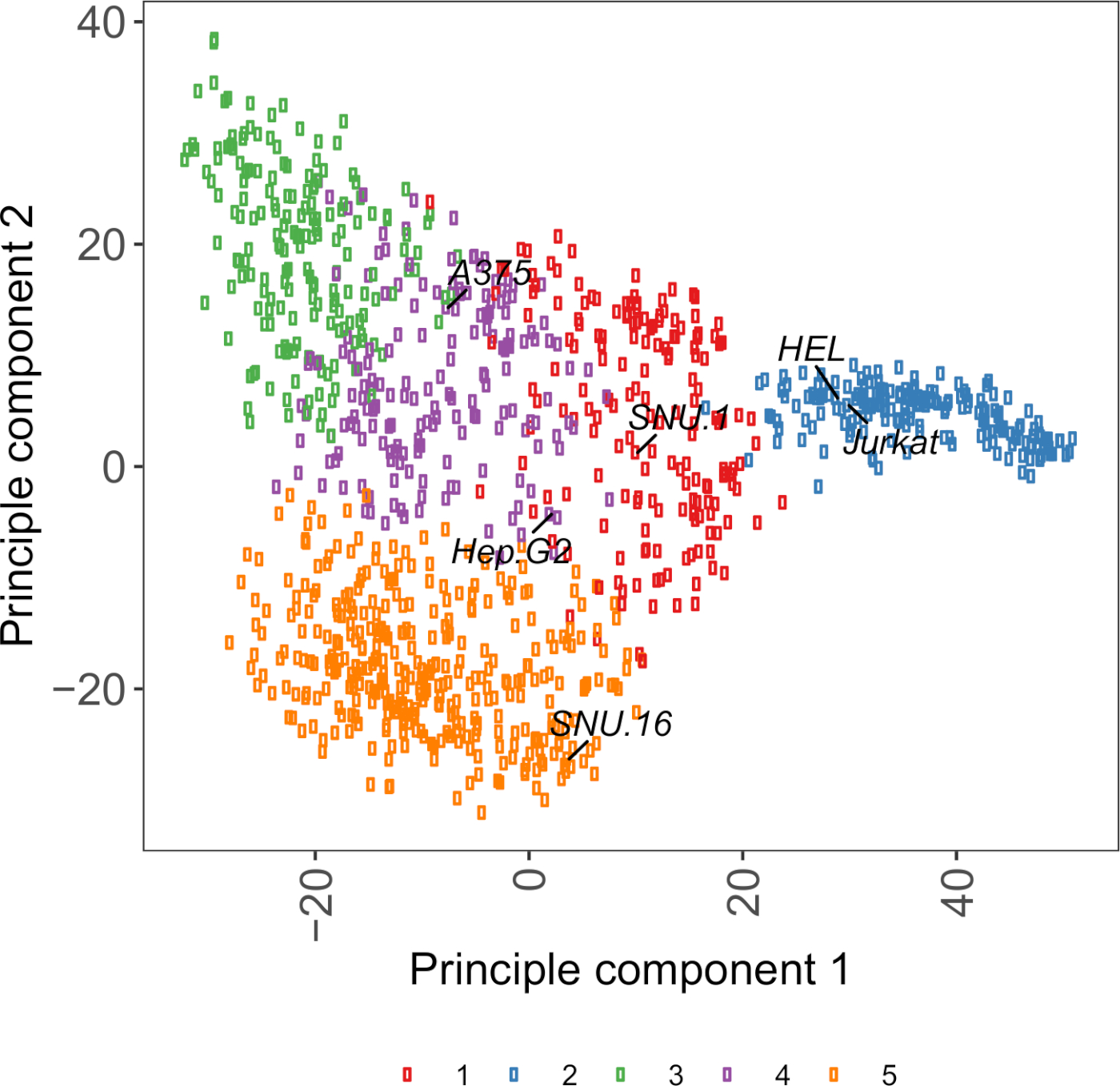
Ergänzende Abbildung S2: Zelloberflächen-Glykoprotein-Transkriptomik-basiertePCA-Diagramm mit RNA-seq-Daten aus über 1.000 Krebszelllinien. Zelllinien aus Cell Model Passport27 wurden mit K-Means-Clustering nach den FPKM-Werten von 1.500 Zell-Oberflächen-Glykoproteinen gruppiert. Repräsentative Zelllinien aus jedem Cluster werden beschriftet. Cluster 5 bestand vollständig aus Zelllinien hämatopoetischen Ursprungs (siehe auch Ergänzende Tabelle 2). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

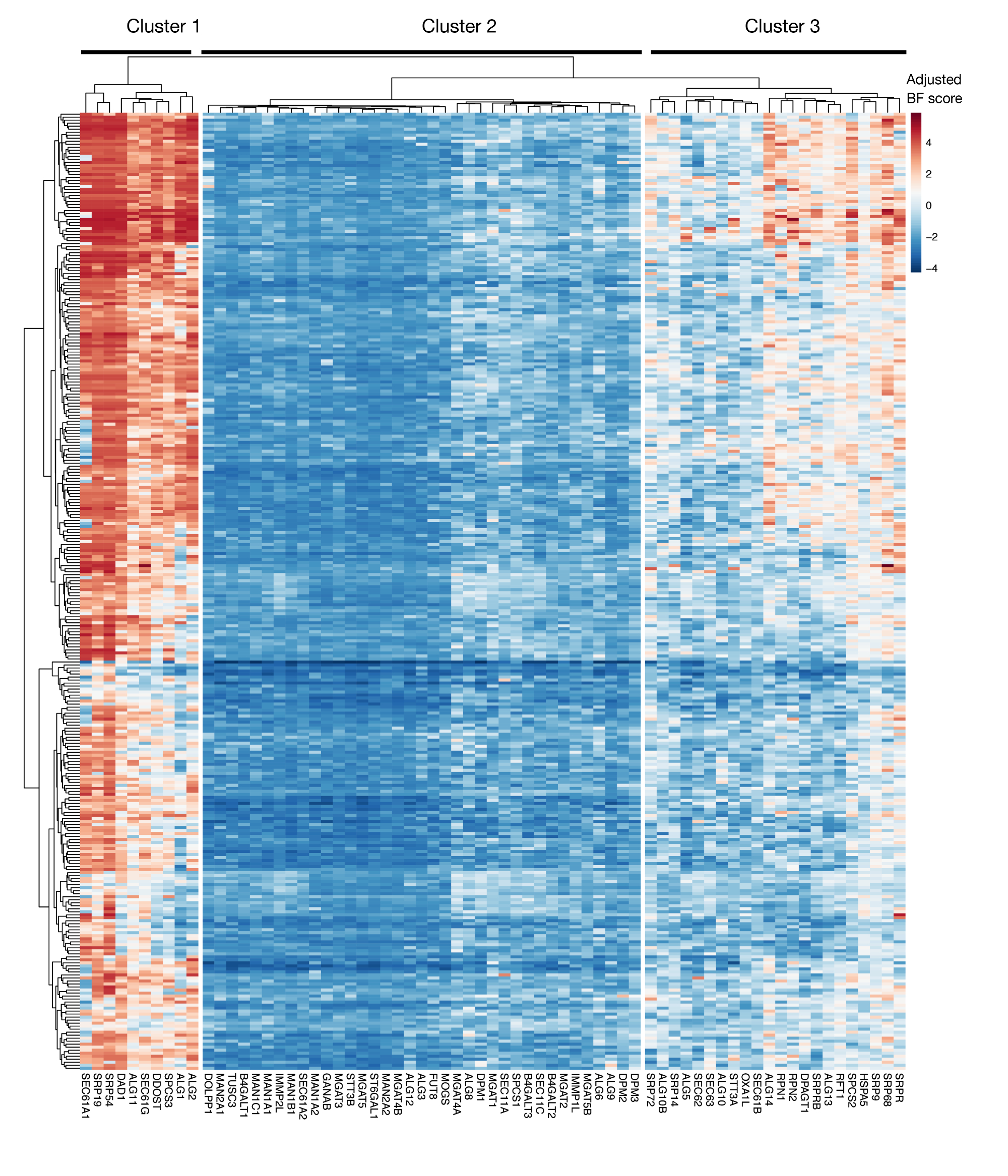
Ergänzende Abbildung S3: Essentialitätswerte für den KEGG-Anmerkungsproteinexport und N-verknüpfte Glykosylationsgene aus Projektergebnissen. Für Gene des Proteinexports und des N-verknüpften Glykosylierungswegs (X-Achse) werden adjusted Bayes-Essentiality-Scores für 330 Zelllinien (Spalten, nicht beschriftet) dargestellt. Werte, die höher als 0 sind, stellen eine signifikante Erschöpfung in der mutierten Population im Vergleich zur ursprünglichen Plasmidbibliothek dar. Die Gene können in drei verschiedene Cluster unterteilt werden, die unterschiedliche Ebenen der Wesentlichkeit in den Zelllinien darstellen. Diese Clusterbildung kann verwendet werden, um den Tag der Sortierung zu bestimmen. Wenn der Bildschirm zu einem späten Zeitpunkt (Tag 16) durchgeführt wird, ist es möglich, dass Gene, die für Zellen als wesentlich bekannt sind (Cluster 1 und 3), nicht identifiziert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergänzende Tabelle 1: Raw count files for and MAGeCK software generated gene_summary files related to the representative genetic screens. Bitte klicken Sie hier, um diese Datei anzuzeigen (Rechtsklick zum Herunterladen).
Ergänzende Tabelle 2: Clustering von Zelllinien nach der Expression von Zelloberflächenrezeptoren. Bitte klicken Sie hier, um diese Datei anzuzeigen (Rechtsklick zum Herunterladen).
Diskussion
Eine CRISPR-basierte Screening-Strategie zur Identifizierung von Genen, die zelluläre Komponenten kodieren, die an der zellulären Erkennung beteiligt sind, wird beschrieben. Ein ähnlicher Ansatz mit CRISPR-Aktivierung bietet auch eine genetische Alternative, um direkt interagierende Rezeptoren von rekombinanten Proteinen zu identifizieren, ohne große Proteinbibliotheken erzeugen zu müssen26. Ein großer Vorteil dieses Ansatzes ist jedoch, dass er auf Wechselwirkungen anwendbar ist, die von Oberflächenmolekülen vermittelt werden, die nativ auf der Zelle angezeigt werden, und nicht von der Überexpression von Rezeptoren abhängt, die die Bindungs-Avidität des Rezeptors beeinflussen können. Im Gegensatz zu anderen Methoden macht diese Technik daher keine Annahmen über die biochemische Natur oder Zellbiologie der Rezeptoren und bietet die Möglichkeit, Wechselwirkungen zu untersuchen, die durch Proteine vermittelt werden, die normalerweise schwierig zu untersuchen sind, indem sie biochemische Ansätze verwenden, wie z. B. sehr große Proteine, oder solche, die die Membran mehrmals durchqueren oder Komplexe mit anderen Proteinen bilden, und Molekülen außer Proteinen wie Glyklyans, Glycolipiden und Phospholipiden. Angesichts der genomskaligen Natur der Methode hat dieser Ansatz auch den Vorteil, nicht nur den Rezeptor zu identifizieren, sondern auch zusätzliche zelluläre Komponenten, die für das Bindungsereignis benötigt werden, und so Einblicke in die Zellbiologie des Rezeptors zu geben.
Eine der hauptwichtigsten Einschränkungen dieser Methode bei der Verwendung von ihm, um den Rezeptor eines verwaisten Proteins zu identifizieren, ist die anfängliche Anforderung, zuerst eine Zelllinie zu identifizieren, die an das Protein bindet. Dies ist nicht immer einfach, und die Identifizierung einer Zelllinie, die einen Bindungsphänotyp anzeigt, der auch für genetische Bildschirme zulässig ist, kann der zeitbegrenzende Schritt für die Bereitstellung dieses Assays sein. Einige Zelllinien neigen dazu, an mehr Proteine zu binden als andere. Dies ist besonders relevant für Proteine, die HS binden, da diese Proteine dazu neigen, an jede Zelllinie zu binden, die HS-Seitenketten anzeigt, unabhängig vom nativen Bindungskontext. Darüber hinaus haben wir beobachtet, dass die Upregulation von Syndecanen (d. h. Proteoglykanen, die HS enthalten) in Zelllinien zu einer erhöhten Bindung von HS-bindenden Proteinen führt26. Dies könnte ein Faktor sein, der bei der Auswahl der Zelllinie für das Screening berücksichtigt werden sollte. Wichtig ist jedoch auch, dass die additive Bindung von HS die Bindung an einen bestimmten Rezeptor nicht beeinträchtigt. Dies bedeutet, dass bei der Bindung es möglich ist, dass es ausschließlich durch HS vermittelt wird, weil die von HS in diesem Test vermittelte Bindung additiv und nicht koabhängig ist19. In einem solchen Szenario kann der beschriebene Heparin-Blockierungsansatz solche Verhaltensweisen identifizieren, ohne einen vollständigen genetischen Bildschirm durchführen zu müssen.
Eine nützliche Ressource für die Auswahl von Zelllinien ist Cell Model Passport, das Genomik, Transkriptomik und Kulturzustandsinformationen für 1.000 Krebszelllinien27enthält. Je nach biologischem Kontext können Zellen anhand ihrer Ausdrucksprofile ausgewählt werden. Um die Auswahl von Zelllinien zu unterstützen, haben wir in Cell Model Passport 1.000 Zellenlinien nach dem Ausdruck von 1.500 vorannotierten menschlichen Zelloberflächenglykoproteinen28 gruppiert (Ergänzende Abbildung 2; Clusterinformationen für jede Zelllinie zusammen mit Wachstumsbedingungen sind in der Ergänzenden Tabelle 2enthalten). Beim Testen der Bindung eines Proteins mit unbekannter Funktion ist es sinnvoll, aus jedem Cluster ein Panel mit repräsentativen Zelllinien auszuwählen, um die Wahrscheinlichkeit zu erhöhen, eine breite Palette von Rezeptoren abzudecken. Bei der Wahl wird empfohlen, Zelllinien zu wählen, die leicht zu beschaffen und leicht zu transduzieren sind. Da diese Zelllinien im Genom-Skalen-Screening verwendet werden, ist es vorzuziehen, dass sie leicht in großen Mengen angebaut werden können und für die lentivirale Transduktion freisind, da es die am häufigsten verfügbare Methode zur Bereitstellung von sgRNA für DAS CRISPR-basierte genetische Screening in den späteren Schritten ist.
Im Allgemeinen werden die Phänotyp-Auswahlen in einer einzigen Sorte durchgeführt. Dies wird jedoch durch die Helligkeit der gefärbten Zellpopulation im Vergleich zur Steuerung bestimmt. Iterative Auswahlrunden könnten für Szenarien angenommen werden, in denen das Signal-Rausch-Verhältnis des gewünschten Phänotyps niedrig ist, oder wenn das Ziel des Bildschirms darin besteht, Mutanten mit starken Phänotypen zu identifizieren. Bei der Verwendung eines iterativen Auswahlansatzes für FACS-basierte Bildschirme ist es wichtig zu berücksichtigen, dass der Sortierprozess zum Zelltod führen kann, hauptsächlich aufgrund der schieren Kraft des Sortierers. Somit werden nicht alle gesammelten Zellen in der nächsten Sortierrunde dargestellt.
Die Komplexität der Bibliothek ist ein sehr wichtiger Faktor bei der Durchführung erfolgreicher genetischer Bildschirme, insbesondere bei negativen Selektionsbildschirmen, da das Ausmaß der Erschöpfung in diesen nur durch einen Vergleich der Ergebnisse mit dem, was in der Startbibliothek vorhanden war, bestimmt werden kann. Bei negativen Auswahlbildschirmen ist es üblich, Bibliotheken mit einer Komplexität von 500-1.000 x zu verwalten. Positive Auswahlbildschirme sind jedoch robuster für Bibliotheksgrößen, da in solchen Bildschirmen nur eine geringe Anzahl von Mutanten für einen bestimmten Phänotyp ausgewählt werden soll. Daher kann im hier beschriebenen positiven Auswahlbildschirm die Bibliotheksgröße auf 50-100x Komplexität reduziert werden, ohne die Qualität des Bildschirms zu beeinträchtigen. Darüber hinaus ist es in diesen Bildschirmen auch möglich, eine Steuerbibliothek für eine bestimmte Zellzeile an einem bestimmten Tag als "allgemeine Kontrolle" für alle Proben zu verwenden, die am Tag für diese angegebene Zelllinie sortiert sind. Dadurch wird die Anzahl der Steuerelementbibliotheken verringert, die erstellt und sequenziert werden müssen.
Eine weitere wichtige Überlegung für die Verwendung dieses Ansatzes sind die Grenzen von Funktionsverlust-Screens bei der Identifizierung von Genen, die für das In-vitro-Zellwachstum unerlässlich sind. Der Zeitpunkt der Bildschirme ist in dieser Hinsicht entscheidend, da je länger die mutierten Zellen in der Kultur gehalten werden, desto höher ist die Wahrscheinlichkeit, dass Zellen mit Mutationen in essentiellen Genen nicht mehr lebensfähig werden und nicht mehr in der mutierten Bibliothek dargestellt werden. Die jüngsten genetischen Screens, die im Rahmen der Project Score-Initiative in über 300 Zelllinien durchgeführt wurden, zeigen, dass mehrere Gene in der KEGG-kommentierten Proteinsekretion und dem N-Glykosylierungsweg oft als wesentlich für eine Reihe von Zelllinien identifiziert werden (Ergänzende Abbildung 3)29. Dies kann bei der Gestaltung von Bildschirmen berücksichtigt werden, wenn die Wirkung von Genen, die für die Proliferation und Lebensfähigkeit erforderlich sind, im Kontext des zellulären Erkennungsprozesses untersucht werden soll. In diesem Fall wäre es generell angemessen, Bildschirme zu einem frühen Zeitpunkt (z. B. Tag 9 Nachtransduktion) durchzuführen. Wenn der Ansatz jedoch verwendet wird, um einige Ziele mit starken Größeneffekten anstelle von allgemeinen zellulären Bahnen zu identifizieren, kann es sinnvoll sein, Bildschirme zu einem späteren Zeitpunkt durchzuführen (z. B. Tag 15-16 Posttransduktion).
Die Ergebnisse des Screenings sind sehr robust; in acht rekombinanten Proteinbindungssieben, die in der Vergangenheit durchgeführt wurden, war der Zelloberflächenrezeptor in jedem Fall der Top-Hit19. Bei der Identifizierung des Interaktionspartners mit diesem Ansatz sollte man daher erwarten, dass der Rezeptor und die Faktoren, die zu seiner Darstellung auf der Oberfläche beitragen, mit einem hohen statistischen Vertrauen identifiziert werden. Sobald der Bildschirm durchgeführt und ein Treffer mit einem einzigen gRNA-Knockout validiert wurde, können weitere Nachuntersuchungen mit vorhandenen biochemischen Methoden wie AVEXIS4 und der direkten sättigungsfähigen Bindung von gereinigten Proteinen mit Oberflächenplasmonresonanz durchgeführt werden. Der hier beschriebene Ansatz gilt für alle Proteine, für die eine lösliche rekombinante Bindungssonde erzeugt werden kann.
Zusammenfassend ist dies ein CRISPR-Knockout-Ansatz im Genommaßstab, um Wechselwirkungen zu identifizieren, die durch Zelloberflächenproteine vermittelt werden. Diese Methode ist allgemein anwendbar, um zelluläre Bahnen zu identifizieren, die für die Zelloberflächenerkennung in einer Vielzahl unterschiedlicher biologischer Kontexte erforderlich sind, einschließlich zwischen den eigenen Zellen eines Organismus (z. B. neuronale und immunologische Erkennung) sowie zwischen Wirtszellen und Pathogenproteinen. Diese Methode bietet eine genetische Alternative zu biochemischen Ansätzen, die für die Identifizierung von Rezeptoren entwickelt wurden, und da sie keine vorherigen Annahmen in Bezug auf die biochemische Natur oder Zellbiologie der Rezeptoren erfordert, hat sie großes Potenzial, völlig unerwartete Entdeckungen zu machen.
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Diese Arbeit wurde durch das Wellcome Trust-Stipendium Nr. 206194 unterstützt, das GJW zugesprochen wurde. Wir danken der Cytometry Core Einrichtung Bee Ling Ng, Jennifer Graham, Sam Thompson und Christopher Hall für die Hilfe bei FACS.
Materialien
| Name | Company | Catalog Number | Comments |
| Anti-mouse alkaline phosphatase | Sigma | A4656 | |
| Blasticidin | Chem-Cruz | SC-204655 | |
| Blood & Cell Culture DNA Maxi Kit | Qiagen | 13362 | |
| BSA | Sigma | A9647-100G | |
| Diethanolamine | Sigma | 398179 | |
| DMEM | Gibco | 31966-021 | |
| Dneasy Blood and Tissue kit | Qiagen | 69504 | |
| DynaMag-96 Side Magnet | Invitrogen | 12331D | |
| HEK293T packaging cells | ATCC | CRL-3216 | |
| Heparin | Sigma | H4784-1G | |
| KAPA HiFi HotStart ReadyMix | Kapa | KK2602 | |
| Lipofectamine LTX with PLUS reagent | Invitrogen | 15338100 | |
| MoFlo XDP cell sorter | BD | ||
| Ni2+-NTA agarose beads | Jena Bioscience | AC-501-25 | |
| OPTI-MEM | Life Technologies | 31985-070 | |
| OX-68 antibody | AbD Serotec | MCA1022R | |
| p-nitrophenyl phosphate | Sigma | 1040-506 | |
| PD-10 desalting columns | GE healthcare | 17085101 | |
| Polybrene | Millipore | TR-1003-G | |
| Polypropylene tubes with 5 mL bed volume | Qiagen | 34964 | |
| Proteinase K, recombinant, PCR Grade | Roche | 3115879001 | |
| Puromycin | Gibco | A11138-03 | |
| Q5 Hot Start High-Fidelity 2× Master Mix | NEB | M0494L | |
| QIAquick PCR purification kit | Qiagen | 28104 | |
| SCFA filter | Nalgene | 190-2545 | |
| Sony Cell sorter | Sony | ||
| SPRI beads (Agencourt AMPure XP beads) | Beckman | A63881 | |
| Streptavidin-coated microtitre plates | Nalgene | 734-1284 | |
| Streptavidin-PE | Biolegend | 405204 |
Referenzen
- Wright, G. J. Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Molecular bioSystems. 5 (12), 1405-1412 (2009).
- van der Merwe, P. A., Barclay, A. N. Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends in Biochemical Sciences. 19 (9), 354-358 (1994).
- Wood, L., Wright, G. J. Approaches to identify extracellular receptor-ligand interactions. Current Opinion in Structural Biology. 56, 28-36 (2019).
- Bushell, K. M., Söllner, C., Schuster-Boeckler, B., Bateman, A., Wright, G. J. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Research. 18 (4), 622-630 (2008).
- Visser, J. J., et al. An extracellular biochemical screen reveals that FLRTs and Unc5s mediate neuronal subtype recognition in the retina. eLife. 4, e08149(2015).
- Özkan, E., et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell. 154 (1), 228-239 (2013).
- Martinez-Martin, N., et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell. 174 (5), 1158-1171 (2018).
- Bianchi, E., Doe, B., Goulding, D., Wright, G. J. Juno is the egg Izumo receptor and is essential for mammalian fertilization. Nature. 508 (7497), 483-487 (2014).
- Mullican, S. E., et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nature Medicine. 23 (10), 1150-1157 (2017).
- Turner, L., et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 498 (7455), 502-505 (2013).
- Frei, A. P., et al. Direct identification of ligand-receptor interactions on living cells and tissues. Nature Biotechnology. 30 (10), 997-1001 (2012).
- Sobotzki, N., et al. HATRIC-based identification of receptors for orphan ligands. Nature Communications. 9 (1), 1519(2018).
- Sharma, S., Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. International Journal of Molecular Sciences. 19 (4), (2018).
- Gebre, M., Nomburg, J. L., Gewurz, B. E. CRISPR-Cas9 Genetic Analysis of Virus-Host Interactions. Viruses. 10 (2), (2018).
- Zotova, A., Zotov, I., Filatov, A., Mazurov, D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. Journal of Immunological Methods. 439, 8-14 (2016).
- Puschnik, A. S., Majzoub, K., Ooi, Y. S., Carette, J. E. A CRISPR toolbox to study virus-host interactions. Nature Reviews. Microbiology. 15 (6), 351-364 (2017).
- Kerr, J. S., Wright, G. J. Avidity-based extracellular interaction screening (AVEXIS) for the scalable detection of low-affinity extracellular receptor-ligand interactions. Journal of Visualized Experiments. (61), e3881(2012).
- Tzelepis, K., et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Reports. 17 (4), 1193-1205 (2016).
- Sharma, S., Bartholdson, S. J., Couch, A. C. M., Yusa, K., Wright, G. J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Research. 28 (9), 1372-1382 (2018).
- Wang, B., et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nature Protocols. 14 (3), 756-780 (2019).
- R Core team. A language and environment for statistical computing. , http://www.R-project.org (2013).
- Hart, T., et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3. 7 (8), 2719-2727 (2017).
- Kuleshov, M. V., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 44 (W1), W90-W97 (2016).
- Crosnier, C., et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 480 (7378), 534-537 (2011).
- Kirk, P., et al. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. The EMBO Journal. 19 (15), 3896-3904 (2000).
- Chong, Z. S., Ohnishi, S., Yusa, K., Wright, G. J. Pooled extracellular receptor-ligand interaction screening using CRISPR activation. Genome Biology. 19 (1), 205(2018).
- van der Meer, D., et al. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Research. 47 (D1), D923-D929 (2019).
- Bausch-Fluck, D., et al. A mass spectrometric-derived cell surface protein atlas. PloS One. 10 (3), e0121314(2015).
- Behan, F. M., et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 568 (7753), 511-516 (2019).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten