
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Obtención de imágenes a largo plazo de poblaciones neuronales identificadas mediante microprismas en animales que se mueven libremente y con la cabeza fija
En este artículo
Resumen
Cuando se integra con una placa de cabeza y un diseño óptico compatible con microscopios de uno y dos fotones, la lente de microprisma presenta una ventaja significativa en la medición de respuestas neuronales en una columna vertical en diversas condiciones, incluidos experimentos bien controlados en estados de cabeza fija o tareas de comportamiento natural en animales que se mueven libremente.
Resumen
Con el avance de la microscopía multifotónica y las tecnologías moleculares, las imágenes de fluorescencia están creciendo rápidamente para convertirse en un enfoque poderoso para estudiar la estructura, la función y la plasticidad de los tejidos cerebrales vivos. En comparación con la electrofisiología convencional, la microscopía de fluorescencia puede capturar la actividad neuronal, así como la morfología de las células, lo que permite registros a largo plazo de las poblaciones de neuronas identificadas con una resolución unicelular o subcelular. Sin embargo, las imágenes de alta resolución generalmente requieren una configuración estable y fija en la cabeza que restringe el movimiento del animal, y la preparación de una superficie plana de vidrio transparente permite la visualización de neuronas en uno o más planos horizontales, pero está limitada en el estudio de los procesos verticales que se ejecutan a diferentes profundidades. Aquí, describimos un procedimiento para combinar una fijación de la placa de la cabeza y un microprisma que proporciona imágenes multicapa y multimodal. Esta preparación quirúrgica no solo da acceso a toda la columna de la corteza visual del ratón, sino que permite obtener imágenes de dos fotones en una posición fija de la cabeza y obtener imágenes de un fotón en un paradigma de movimiento libre. Con este enfoque, se pueden muestrear poblaciones celulares identificadas en diferentes capas corticales, registrar sus respuestas en estados fijos y de movimiento libre, y realizar un seguimiento de los cambios a largo plazo durante meses. Por lo tanto, este método proporciona un ensayo completo de los microcircuitos, lo que permite la comparación directa de las actividades neuronales evocadas por estímulos bien controlados y bajo un paradigma de comportamiento natural.
Introducción
El advenimiento de las imágenes fluorescentes de dos fotones in vivo 1,2, que combinan las nuevas tecnologías en sistemas ópticos e indicadores de fluorescencia modificados genéticamente, ha surgido como una técnica poderosa en neurociencia para investigar la intrincada estructura, función y plasticidad en el cerebro vivo 3,4. En particular, esta modalidad de imagen ofrece una ventaja sin precedentes sobre la electrofisiología tradicional al capturar tanto la morfología como las actividades dinámicas de las neuronas, lo que facilita el seguimiento a largo plazo de las neuronas identificadas 5,6,7,8.
A pesar de sus notables fortalezas, la aplicación de imágenes de fluorescencia de alta resolución a menudo requiere una configuración estática fija de la cabeza que restringe la movilidad del animal 9,10,11. Además, el uso de una superficie de vidrio transparente para visualizar neuronas restringe las observaciones a uno o más planos horizontales, lo que limita la exploración de la dinámica de los procesos verticales que se extienden a través de diferentes profundidades corticales12.
Abordando estas limitaciones, el presente estudio describe un procedimiento quirúrgico innovador que integra la fijación de la placa de la cabeza, el microprisma y el miniscopio para crear una modalidad de imagen con capacidades multicapa y multimodal. El microprisma permite observar el procesamiento vertical a lo largo de la columna cortical 13,14,15,16, lo cual es fundamental para comprender cómo se procesa y transforma la información a medida que se mueve a través de las diferentes capas de la corteza y cómo se altera el procesamiento vertical durante los cambios plásticos. Además, permite obtener imágenes de las mismas poblaciones neuronales en un paradigma de fijación de la cabeza y en un entorno de movimiento libre, que abarca los versátiles entornos experimentales 17,18,19: por ejemplo, la fijación de la cabeza a menudo se requiere para paradigmas bien controlados como la evaluación de la percepción sensorial y los registros estables bajo el paradigma de 2 fotones, mientras que el movimiento libre ofrece un entorno más natural y flexible para los estudios del comportamiento. Por lo tanto, la capacidad de realizar una comparación directa en ambos modos es crucial para avanzar en nuestra comprensión de los microcircuitos que permiten respuestas flexibles y funcionales.
En esencia, la integración de la fijación de la placa de la cabeza, el microprisma y el miniscopio en las imágenes de fluorescencia ofrece una plataforma prometedora para sondear las complejidades de la estructura y funcionalidad del cerebro. Los investigadores pueden tomar muestras de poblaciones celulares identificadas a través de varias profundidades que abarcan todas las capas corticales, comparar directamente sus respuestas tanto en paradigmas bien controlados como naturales, y monitorear sus alteraciones a largo plazodurante meses. Este enfoque ofrece información valiosa sobre cómo estas poblaciones neuronales interactúan y cambian a lo largo del tiempo en diferentes condiciones experimentales, proporcionando una ventana a la naturaleza dinámica de los circuitos neuronales.
Protocolo
Todos los experimentos se llevaron a cabo de acuerdo con la Ley de Animales (Procedimientos Científicos) del Reino Unido de 1986 bajo licencias personales y de proyectos aprobadas y emitidas por el Ministerio del Interior del Reino Unido después de la revisión ética correspondiente. Líneas transgénicas adultas CaMKII-TTA; GCaMP6S-TRE21 fueron criados y sus descendientes utilizados en el experimento. Para la seguridad de los experimentadores y el mantenimiento de las condiciones estériles, todos los procedimientos se realizaron en condiciones asépticas y con equipos de protección personal completos.
1. Preparación preoperatoria
- Para minimizar el edema, administrar dexametasona (0,2 mg/kg) por vía subcutánea, 12-24 h antes de la cirugía.
- Esterilice todos los instrumentos quirúrgicos en un autoclave y esterilice el área quirúrgica con ácido hipocloroso estabilizado con agua destilada y etanol al 70% antes de la cirugía. Asegúrese de que todo el equipo quirúrgico esté encendido.
- Anestesiar al animal (24 semanas de edad, macho de 31 g de peso) con isoflurano con una dosis de inducción del 5%, que se reduce al 1%-2% una vez que el ratón está en el marco estereotáxico, con O2 mantenido entre 1-2 L/min. Inyectar AINE (Carprofeno, 2,5 mg/kg) por vía subcutánea.
- Verifique la ausencia de reflejo de pellizco de los dedos de los pies para evaluar la profundidad de la anestesia (aumente la concentración de isoflurano en incrementos del 0,5% si se observa reflejo).
- Afeita la cabeza del animal, con una recortadora, desde detrás de las orejas hasta ligeramente por encima de los ojos. Limpie esta área con una toallita con alcohol y una solución de povidona yodada, asegurándose de evitar el contacto con los ojos del animal.
- Monta al animal en la almohadilla térmica homeotérmica y en el marco estereotáxico equipado con barras para las orejas y los dientes y asegura la cabeza. Asegúrese de que la cabeza esté estable, ya que esto es crucial para que el siguiente procedimiento sea exitoso (Figura 1).
- Aplique ungüento oftálmico sobre los ojos del animal para evitar que se sequen durante la cirugía y cúbralos con papel de aluminio para protegerlos de la luz. Cubra al animal con una funda quirúrgica estéril.

Figura 1: Preparación preoperatoria. El ratón se coloca en el marco estereotáxico, asegurado por una pieza nasal y barras en las orejas. El ratón se coloca sobre una almohadilla térmica con temperatura regulada. Los ojos tienen ungüento oftálmico y están cubiertos por papel de aluminio. Se afeita la cabeza y se expone el cráneo. Se coloca una funda estéril sobre el animal. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
2. Craneotomía
- Con unas tijeras quirúrgicas, haga una incisión en la piel a lo largo de la línea media del área afeitada de la cabeza para exponer el cráneo.
- Limpie el cráneo con un hisopo de algodón estéril y peróxido de hidrógeno diluido (3% p/v de 35% H2O2 en 97% dH2O) durante 1-3 s para eliminar cualquier tejido conectivo (Figura 2A). Seque el cráneo con una gota de etanol al 70% y un nuevo hisopo de algodón estéril.
- Alinee el cráneo anterior/posterior (AP) y medial/lateral (ML) para garantizar un sitio de implantación preciso. Para ello, mida la profundidad dorsoventral (DV) del cráneo tanto en el bregma como en la lambda y asegúrese de que la diferencia entre ambos sea de <0,03 mm. Para la alineación mediolateral, mida los puntos equidistantes en ambos huesos parietales desde la línea media y nuevamente asegúrese de que la diferencia de VD sea de <0,03 mm.
- Usando bregma como origen, encuentre y marque el área cortical deseada; en este caso, se trata de la corteza visual primaria monocular (V1), AP: -3,5 mm, ML: -2,5 mm.
- Utilice una broca de trépano (1,8 mm de diámetro) y una fresa dental (velocidad de 10.000 rpm) para exponer la corteza, asegurándose de que la marca de la zona cortical deseada (monocular V1) se encuentre dentro del tercio inferior de la ventana de la broca.
- Asegúrese de que el ángulo de la broca sea perpendicular a la curvatura del cráneo. Esto asegurará una craneotomía uniforme y evitará daños en la duramadre o la corteza.
- Taladre hasta que haya una disminución en la resistencia y luego deténgase (Figura 2B). Retire con cuidado el fragmento óseo desprendido con la punta de una aguja 23G (Figura 2C).
- Limpie la corteza expuesta con espuma quirúrgica saturada en líquido cefalorraquídeo artificial frío (ACSF, por sus siglas en inglés) para eliminar cualquier residuo y detener cualquier sangrado que pueda ocurrir.
- Mantenga siempre hidratada la corteza expuesta, utilizando ACSF frío, durante toda la cirugía.

Figura 2: Craneotomía. (A) Se muestra una incisión en la piel entre el bregma y la lambda. Se ha extirpado tejido conectivo de la superficie expuesta. (B) Craneotomía mediante fresa de trépano antes de la extracción del fragmento óseo. (C) Craneotomía después de la extracción del fragmento óseo, que muestra la duramadre y la corteza intactas (la barra de escala representa 0,5 mm). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
3. Incisión precortada
NOTA: Para ser considerado al realizar la incisión precortada, la incisión y la implantación del microprisma deberán ser anteriores a la región de interés (ROI) de la imagen. Esto es para permitir un campo de visión completo y preciso. En el contexto de este protocolo, la incisión se realizará a lo largo del eje mediolateral, y el microprisma se orientará hacia la parte posterior (Figura 3B).
- Para ayudar con la inserción y aliviar la presión en la corteza durante la inserción del microprisma, haga una incisión.
- Coloque el bisturí quirúrgico en el soporte del brazo estereotáxico y oriente la hoja o el brazo estereotáxico de modo que corte a lo largo del eje ML.
- Mueva la cuchilla a la coordenada AP deseada (AP: -3,4 mm); se requiere que el prisma esté delante de la ROI, por lo que la incisión debe ser 100 μm anterior a la coordenada AP de la ROI de la imagen (-3,5 mm).
- Ahora moviendo el bisturí hacia el borde medial de la craneotomía, donde se encuentra con el cráneo, baje lentamente el bisturí hasta que llegue al hueso y luego deténgase. Como el grosor óseo es de 200 μm, incorpore este valor a la profundidad total de inserción (consulte el cálculo del paso 5.1).
- La imagen óptima está en el centro del prisma, es decir, 500 μm; por lo tanto, asegúrese de que esta profundidad se alinee con la profundidad de la columna cortical (ROI DV: - 0,35 mm).
- Incorporando el grosor del cráneo en el cálculo de la profundidad, use la siguiente ecuación, que determina qué tan profunda debe ser la incisión precortada desde la superficie del cráneo. Para este protocolo, la profundidad de implantación se calcula como:
Grosor óseo (200 μm) + ROI de imagen (p. ej., 350 μm) + profundidad de microprisma restante (500 μm) = 1.050 μm
- Incorporando el grosor del cráneo en el cálculo de la profundidad, use la siguiente ecuación, que determina qué tan profunda debe ser la incisión precortada desde la superficie del cráneo. Para este protocolo, la profundidad de implantación se calcula como:
- Asegúrese de que la longitud de la incisión sea superior a 1 mm, pero no excesiva; por lo tanto, una distancia de 1,2 mm es ideal, con la coordenada monocular ML en el medio de esta distancia.
- Cuando esté listo para realizar la incisión, retire el exceso de ACSF para que la visión no se oscurezca (Figura 3A).
- Mueva el bisturí desde el borde medial de la craneotomía hasta la coordenada medial inicial de la incisión. Baje lentamente (10 μm/s) el cuchillo hacia la corteza.
- Una vez perforada la duramadre y el cuchillo ha entrado en la corteza, aplique una gota de ACSF frío sobre la corteza para mantener el tejido lubricado e hidratado durante la incisión.
- Una vez alcanzada la profundidad final, comience a mover la cuchilla a lo largo del eje ML (a una velocidad de 10 μm/s).
- Siga observando el tejido circundante a medida que se realiza la incisión. Si el pañuelo se arrastra junto con el cuchillo, muévalo hacia arriba y hacia abajo unas cuantas veces para asegurarse de que se esté cortando el pañuelo, y luego continúe lateralmente, recordando volver a colocar el cuchillo en su profundidad final.
- Cuando haya terminado, levante lentamente el cuchillo. Si sale sangre durante la incisión, use este tiempo para limpiar el sitio de la incisión con espuma quirúrgica empapada en ACSF para diluir la sangre y empujar la sangre dentro de la incisión hacia afuera. Recuerde dejar una espuma quirúrgica fresca y saturada sobre la corteza expuesta hasta que esté lista para insertar el microprisma.

Figura 3: Implantación del microprisma. (A) Incisión precortada. (B) Esquema de la lente de microprisma integrada que demuestra su posición dentro de la corteza (C) Lente de microprisma integrada en la orientación correcta para la incisión precortada antes de la inserción en la corteza (la barra de escala representa 0,5 mm). (D) Ejemplo de acumulación de cemento alrededor de la lente integrada para asegurar su fijación al cráneo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
4. Inserción de microprismas e implantación de la placa de la cabeza
- La lente de microprisma está compuesta por una lente de índice degradado unida a un prisma en el extremo distal de la lente, que está integrado en una placa base. Coloque el microprisma en el kit de implantes.
- Asegúrese de que el lado de imagen del prisma esté opuesto al tornillo de la placa base. Para ayudar con la inserción y colocación del microprisma, fíjelo al marco estereotáxico y oriente el prisma de modo que se alinee con la incisión (Figura 3C).
- Baje lentamente el microprisma en el sitio de la incisión (10 μm/s). Recuerde quitar el ACSF cuando inserte inicialmente el prisma, pero una vez en la corteza, enjuague con ACSF frío para lubricar la inserción.
- La corteza debe permanecer estable mientras el prisma se baja en la incisión; si no es así, agregue ACSF adicional y agite la corteza moviendo el prisma hacia arriba y hacia abajo para aflojar la corteza del prisma.
- Una vez alcanzada la profundidad final, secar la superficie cortical expuesta con un tejido estéril, teniendo cuidado de no tocar el prisma.
- Cubra el área cortical expuesta que rodea el prisma, así como la lente, con una capa protectora de adhesivo de silicona, minimizando el exceso de adhesivo en el cráneo circundante y el telescopio ficticio.
- Una vez curado (5-10 min), coloque la placa de la cabeza en el cráneo para estabilizar la cabeza durante la toma de imágenes fijas de la cabeza.
- Asegúrese de que la placa de cabeza sea lo suficientemente posterior para no interferir con la colocación del implante y permitir la aplicación adecuada de cemento para asegurar adecuadamente el implante.
- Asegúrese de que la línea media de la placa frontal se encuentre ligeramente a la derecha de la lente implantada para asegurarse de que ambos lados de la placa frontal puedan fijarse a la platina de la cabeza cuando realice experimentos con la cabeza fija
- Aplique cemento adhesivo en la placa de la cabeza y el cráneo.
- Prepare el cemento dental adhesivo mezclando 1 cucharada de polvo de cemento opaco con 4 gotas de medio de mezcla y aplicando una gota del catalizador.
- Coloque cemento tanto en la placa de la cabeza como en el cráneo y mantenga la placa de la cabeza en su lugar hasta que se cure, asegurándose de que esté paralela a las barras de las orejas (a través de un examen visual, inspeccione tanto desde arriba como desde detrás de la cabeza del animal).
- Aplique cemento adhesivo para cubrir el resto del cráneo y el tejido expuestos, incorporando el microprisma (hasta la base de la placa base) y la placa de cabeza.
- No deje que el cemento entre en contacto con la placa base, el microscopio ficticio ni ninguno de sus componentes. Siga aplicando el cemento adhesivo hasta que el microprisma y la placa de cabeza estén cubiertos y estables (Figura 3D).
- Cuando el cemento se haya curado, separe el microscopio ficticio moviendo lentamente el brazo estereotáxico hacia arriba mientras estabiliza el microprisma con pinzas (están conectadas a través de imanes, por lo que se puede sentir cierta resistencia durante la separación).
- Inserte la cubierta protectora en la lente y apriete el tornillo para asegurarla en su lugar.
- Retirar al animal del marco estereotáxico, dejar que se recupere en una caja de recuperación tibia y administrar solución salina estéril tibia al 0,9% por vía subcutánea (3% del peso corporal).
- Una vez que el animal esté despierto y en movimiento, colóquelo de nuevo en una jaula limpia de una sola casa. Monitorear al animal y administrar analgesia postoperatoria adicional de acuerdo con la política de analgesia de la institución local.
- Espere 4 semanas después de la cirugía, el animal debe estar listo para la toma de imágenes.
5. Imágenes de calcio de un fotón de capas corticales en ratones que se mueven libremente
NOTA: Es esencial utilizar las imágenes capturadas de la sesión de imágenes original cada vez para garantizar una adquisición precisa del plano de imagen deseado. Estos puntos de referencia identificados, junto con las neuronas, desempeñan un papel fundamental en el proceso de alineación descrito en detalle en el paso 9 del protocolo. Al adquirir datos de un fotón, el miniscopio es tanto el sistema de imágenes como la fuente láser. La excitación utiliza LED con un rango de potencia de 0-2 mW/mm2 en la superficie frontal del objetivo. El láser utiliza una longitud de onda de excitación de 455 ± 8 nm (luz azul) para la señalización GCaMP. El control deslizante de enfoque de la lente se puede usar para ajustar el enfoque (eje Z), que se representa en la interfaz como 0-1000, donde 0 representa una distancia de trabajo de 0 μm y 1000 representa la distancia de trabajo máxima de 300 μm.
- Antes de adquirir datos, deje que el animal se aclimate a la habitación y a la arena abierta durante 1 hora antes de la sesión de grabación.
- Antes de la toma de imágenes, desinfecte y limpie todo con desinfectantes apropiados (p. ej., ácido hipocloroso estabilizado con agua destilada y etanol al 70%).
- Configure la caja DAQ conectándola a una computadora e iniciando el software de adquisición de datos. Establezca una conexión directa a través de un cable ethernet para minimizar la caída de tramas; Sin embargo, el modo de conexión inalámbrica puede ser suficiente dependiendo de la intensidad de la conexión inalámbrica.
- Coloque el miniscopio en la placa base del animal bajo un pescuezo suave.
- Primero, retire la cubierta protectora de la placa base desenroscando el tornillo de fijación. Sujete la cubierta por su abertura con pinzas. A continuación, coloque el miniscopio en la placa base, donde se asienta la cubierta.
- Compruebe la orientación del miniscopio en relación con la placa base antes de instalarlo, de modo que el lado con la marca del tornillo mire hacia el tornillo.
- Una vez que el miniscopio esté colocado, apriete el tornillo de fijación para estabilizarlo. Solo avance el tornillo de fijación hasta que se pueda sentir algo de resistencia. Apretar demasiado el tornillo de fijación podría dañar el miniscopio y, por lo tanto, debe evitarse.
- Conecte el miniscopio a la caja DAQ y prepare el software para la grabación.
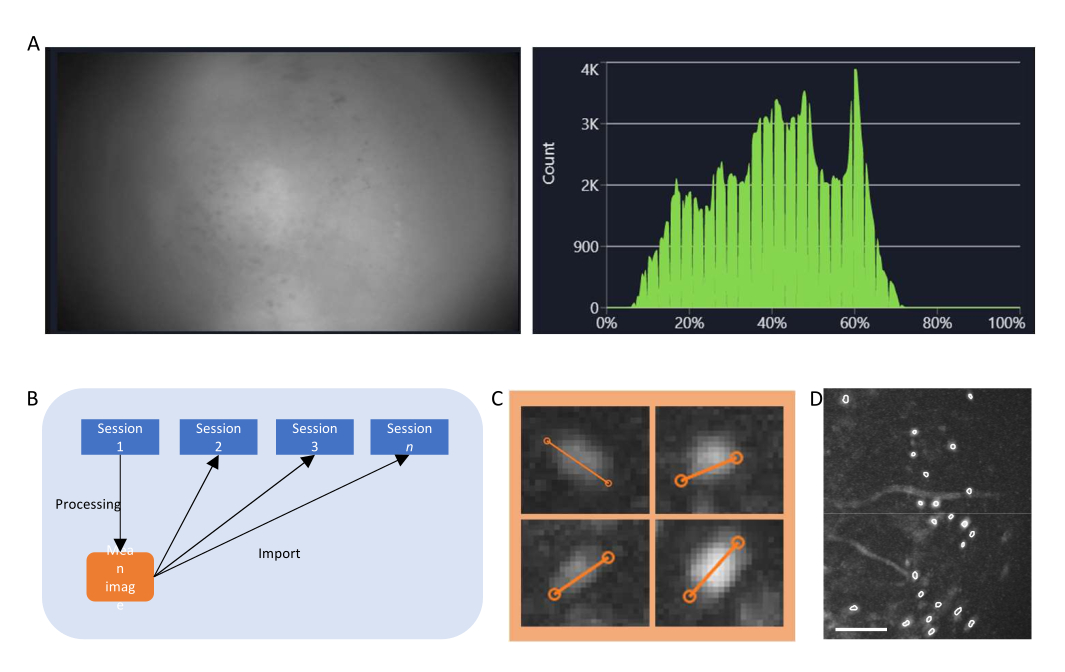
- En el software de adquisición de datos, encienda la transmisión del miniscopio y ajuste los parámetros de grabación (velocidad de fotogramas de imagen, ganancia, potencia del LED, valor de enfoque electrónico) para lograr un campo de visión claro (Figura 4A).
- Encienda la ventana del histograma y ajuste la ganancia y la potencia del LED para que la intensidad registrada se sitúe entre el 35% y el 70%.
- Si va a realizar un estudio longitudinal, consulte las grabaciones o instantáneas tomadas en una sesión anterior y ajuste el valor de efocus para que se vea claramente el mismo plano de imagen.
- Inicie el experimento y la adquisición de datos.
- Después de la finalización del experimento, retire al animal del aparato conductual.
- Bajo un suave pescuezo, afloje el tornillo de fijación y retire el miniscopio de la placa base del animal.
- Vuelva a colocar la cubierta protectora en la placa base y estabilícela con el tornillo de fijación.
- Regrese al animal a su jaula de origen (continúe con el paso 7 si desea procesar datos de un fotón).

Figura 4: Adquisición y procesamiento de datos con software. (A) Una imagen que muestra la transmisión en tiempo real desde el miniscopio. Se recomienda ajustar el valor de enfoque de la lente, de modo que se vea una vista clara en la ventana de transmisión, junto con la ganancia y la potencia del láser de imagen (B) Gráfico esquemático que ilustra el flujo de trabajo de alineación recomendado para sesiones grabadas en diferentes puntos de tiempo. Se recomienda generar una imagen media desde la primera sesión, siguiendo las instrucciones del software de procesamiento de datos. Esta imagen debe utilizarse como imagen de referencia durante la corrección de movimiento para las siguientes sesiones. (C) Ejemplos de cuatro celdas de la misma imagen ΔF/F proyectada al máximo. Se dibuja una línea naranja a través de cada celda para medir el diámetro de su celda en píxeles, cuyo promedio se toma como argumento de entrada para el algoritmo de identificación de celdas (arriba a la izquierda: 13, arriba a la derecha: 11, abajo a la izquierda: 12, abajo a la derecha: 13). (D) Salida del algoritmo de identificación de celdas después de la curación manual (imagen recortada). Los contornos blancos representan las celdas identificadas (la barra de escala representa 100 μm). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
6. Imágenes de calcio de dos fotones de capas corticales en ratones fijados a la cabeza
NOTA: Para la microscopía de barrido láser de dos fotones, la fuente de luz es un láser ultrarrápido sintonizable con una longitud de onda de excitación de 920 nm. La potencia de excitación, medida en el objetivo, era típicamente entre 100-150 mW y se ajustaba en cada sesión para lograr niveles similares de fluorescencia. La luz de emisión se filtró mediante un filtro de emisión (525/70 nm) y se midió mediante un tubo fotomultiplicador independiente (PMT), denominado canal verde. Las imágenes se adquirieron con un objetivo de inmersión en aire de 20x (NA = 0,45, distancia de trabajo de 6,9-8,2 mm).
- Antes de adquirir datos, habitúe al animal al aparato en los días previos (para que el animal se sienta cómodo con la configuración del comportamiento). Deje que el animal pase de 15 a 30 minutos al día, durante 2 a 3 días, explorando la configuración o hasta que muestre un comportamiento naturalista antes de comenzar la adquisición de datos.
- Encienda el sistema de imágenes de dos fotones, inicie el software de adquisición y encienda el láser. Asegúrese de que los láseres estén obturados y que los PMT estén apagados antes de continuar.
- Limpie el aparato de imágenes de dos fotones con ácido hipocloroso estabilizado con agua destilada y etanol al 70%.
- Asegúrese de ajustar el aparato para que se ajuste al tamaño del animal. Coloque suavemente el mouse y su placa frontal en la configuración de la etapa de la cabeza y atorníllela en su lugar para estabilizar la cabeza del mouse.
- Una vez colocado, retire la cubierta de la lente (consulte el paso 5.4.1) y alinee el objetivo de modo que quede sobre la placa base.
- Utilice los controles de epifluorescencia y de la etapa XYZ para enfocar el tejido cortical.
- Una vez que las capas corticales sean visibles, cambie el microscopio para permitir la obtención de imágenes de dos fotones (cambie el espejo por el espejo dicroico, cierre el obturador fluorescente y apague el láser de epifluorescencia y el monitor). Asegúrese de apagar las luces principales para proteger los PMT cuando tome imágenes.
- Configure parámetros para optimizar los archivos de imagen adquiridos.
- Utilice el modo de adquisición resonante para obtener imágenes de calcio, ya que puede capturar el disparo rápido de señales GCaMP.
- Ajuste la potencia del láser, la ganancia PMT, el zoom y las tablas de búsqueda (LUT) para obtener una imagen óptima y consulte la imagen de un fotón para asegurarse de que se está fotografiando el plano focal correcto.
- Comience a obtener imágenes de las capas corticales con el sistema de dos fotones con monitoreo sincronizado del comportamiento y entradas de estímulos (si corresponde).
- Asegúrese de guardar estos parámetros de adquisición y distancias XYZ si desea reproducir imágenes idénticas a lo largo del tiempo.
- Para capturar una pila z de las capas corticales, siga los pasos que se describen a continuación.
- Encuentre el plano desde el que iniciar la pila z, ajuste los parámetros de adquisición para optimizar la imagen y márquelo como punto de partida en el software.
- A continuación, usando el control Z, muévase hacia abajo en la pila, ajustando solo la potencia del láser para mantener una luminancia constante de la pila, y marque el final de la pila en el software.
- Paso crítico: usando la opción Gradiente exponencial relativo en la pestaña Gradiente de potencia láser , permita que el software calcule el aumento de la potencia láser a medida que se mueve a través de la pila z. Asegúrese de anotar los valores de potencia del láser del punto final en la tabla proporcionada por el software para permitirle calcular el gradiente.
- Una vez establecidos los parámetros de la pila z, ajuste el tamaño del paso (μm).
NOTA: El tamaño del paso determinará el tiempo empleado, el número de sectores y la calidad de detalle de la pila. Los tamaños de paso más pequeños darán como resultado un tiempo de adquisición más largo, un aumento en el número de sectores y un mejor detalle en comparación con un tamaño de paso más grande. Las pilas Z se utilizan para ayudar con el registro de imágenes de uno y dos fotones, ya que resaltarán cualquier punto de referencia o característica anatómica.
- Para adquirir una serie temporal (serie T) de cambios de calcio en las neuronas, encuentre un plano focal óptimo utilizando los controles de la platina XYZ y ajuste la potencia del láser, la ganancia PMT, el zoom y las LUT.
- En la pestaña Serie T del software de adquisición, defina los parámetros de frecuencia de adquisición para que coincidan con los datos adquiridos mediante el sistema de imágenes de un fotón.
NOTA: La coincidencia de la frecuencia hará que los datos 1P y 2P sean comparables, y es más adecuado para el algoritmo de identificación de celdas utilizado en los pasos de procesamiento de datos. Se pueden incorporar múltiples disparadores y otros modos de adquisición en la adquisición de la serie T.
- En la pestaña Serie T del software de adquisición, defina los parámetros de frecuencia de adquisición para que coincidan con los datos adquiridos mediante el sistema de imágenes de un fotón.
- Comienza la adquisición de la serie T.
7. Procesamiento de datos de imágenes de calcio de un fotón
- Para grabar vídeos con un fotón, utilice el software de procesamiento de datos que se incluye con el sistema de miniscopio.
- En primer lugar, preprocese la película mediante un muestreo espacial y temporal. Por lo general, la reducción de la resolución espacial de la película en un factor de dos reduciría significativamente el tiempo de procesamiento sin comprometer gravemente la precisión de la identificación de células.
- Establezca el factor de reducción de muestreo temporal para que la velocidad de fotogramas de la película se reduzca a unos 10 Hz, que es más adecuado para el algoritmo de identificación de celdas utilizado en los pasos siguientes.
- Si adquiere varias películas en el mismo día de la imagen, combine las películas en una serie temporal antes del paso de preprocesamiento para procesarlas juntas.
- Opcional: Aplique un filtro de paso de banda espacial a la película para eliminar las frecuencias espaciales más bajas y más altas, lo que da como resultado una película más suave con mayor contraste.
- Registre la película utilizando la función de corrección de movimiento del software. De este modo, se registra la película y se corrigen los artefactos de movimiento causados por el movimiento del miniscopio en relación con la superficie de la imagen.
- Paso crítico: Si se realiza un estudio longitudinal, registre las películas en el mismo campo de visión, por ejemplo, la imagen media de la película tomada el primer día de obtención de imágenes (Figura 4B).
- Calcule el ΔF/F de la película utilizando la pestaña correspondiente y proyecte la película para generar una imagen de proyección máxima de la película ΔF/F. Esta imagen mostrará regiones que muestran cambios en los niveles de fluorescencia, potencialmente neuronas individuales, y podría usarse para medir el diámetro promedio de las neuronas (Figura 4C).
- Alternativamente, mida el diámetro de la célula en la película corregida por movimiento, donde las neuronas muestran una fluorescencia clara.
- Identifique las células utilizando los algoritmos del software.
- Si bien hay dos opciones disponibles (PCA-ICA y CNMF-E) en este paso, utilice CNMF-E para este estudio. Introduzca el diámetro medio de la celda en píxeles y ejecute el algoritmo para generar un conjunto de celdas que contenga regiones de interés (ROI) que demuestren actividades similares a las de las células.
- Seleccione manualmente las ROI que son células (tienen una morfología similar a la de una célula y una actividad22,23, y se encuentran dentro del campo de visión) de las que no lo son, y valide el conjunto de células seleccionadas (Figura 4D).
- Exporte las trazas de calcio de cada ROI para su posterior análisis.
8. Procesamiento de datos de imágenes de calcio de dos fotones
- Para películas de grabación de dos fotones, utilice un paquete de Python diseñado para procesar datos de análisis de calcio de dos fotones.
- En primer lugar, combine las imágenes tomadas en una serie T en una pila .tiff como se describe a continuación.
- En la interfaz Opciones de ejecución , ajuste los parámetros, incluido el valor tau y la velocidad de fotogramas, para que coincida con el GCaMP utilizado y la velocidad de fotogramas de la grabación.
- Opcional: Establezca el parámetro do_registration en 1 para registrar la película. Esto es equivalente al paso de corrección de movimiento descrito anteriormente.
- Opcional: Establezca el parámetro anatomical_only en 1 para detectar ROI utilizando las características anatómicas además de la dinámica de fluorescencia. Esto requiere la entrada del diámetro de la celda, así que tome medidas utilizando el software de procesamiento de imágenes. Por lo general, esto se recomienda, ya que genera ROI con formas más naturales (Figura 5A-C).
- Una vez que se hayan establecido todos los parámetros, ejecute el algoritmo para que realice todos los cálculos juntos. Consulte la interfaz gráfica de usuario (GUI) para comprobar el progreso.
- Una vez hecho esto, vuelva a la interfaz de selección de celdas para la selección manual de los resultados de identificación de celdas.
- Guarde una imagen del conjunto de celdas seleccionadas en la parte superior de la proyección máxima de la película. Esto se utilizará más adelante como imagen de referencia para registrar datos de registro de un fotón.
- A continuación, el algoritmo guarda los resultados automáticamente. npy, al que se puede acceder más adelante con Python. Alternativamente, guarde los resultados en otros archivos formateados para su posterior análisis en otro software.

Figura 5: Identificación de células mediante software de procesamiento de dos fotones. (A) Imagen representativa de la identificación de células tomada del software de procesamiento de dos fotones. Al establecer Anatomical_only parámetro en 0 pero manteniendo todos los demás parámetros iguales, hay varias no-celdas presentes en el área entre las líneas discontinuas que interfieren con la curación manual de las celdas reales. (B) Ejemplos de mediciones del diámetro de la célula tomadas de (A), utilizando un software de procesamiento de imágenes (arriba a la izquierda; 7,5 píxeles, arriba a la derecha; 9, abajo a la izquierda; 6,5, abajo a la derecha; 7,5). (C) Imagen representativa de la identificación celular. Al establecer Anatomical_only parámetro en 1 e introducir el diámetro medio de la célula tomado de (B) en el algoritmo de diámetro de la célula, no hay células presentes en el área entre las líneas discontinuas (las barras de escala representan 200 μm). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
9. Registro de conjuntos celulares identificados en todas las modalidades de imagen
- Llevar a cabo el registro de células identificadas a partir de registros de un fotón y dos fotones con el algoritmo de registro y análisis de imágenes multimodales (MIRA), que está disponible a través de la interfaz Python del software de imágenes de un fotón.
- Este algoritmo alinea los datos de un fotón y dos fotones a través de un registro no rígido. Vea el conjunto de cuadernos de demostración en línea que se encuentran en el sitio web y que se utilizaron para este estudio.
NOTA: Los cuadernos se escribieron para que todo el procesamiento se completara en el software 1P y, por lo tanto, no son compatibles con el software de procesamiento 2P. Por lo tanto, solo siga algunos de los pasos de los cuadernos para este estudio.
- Este algoritmo alinea los datos de un fotón y dos fotones a través de un registro no rígido. Vea el conjunto de cuadernos de demostración en línea que se encuentran en el sitio web y que se utilizaron para este estudio.
- Siga los pasos descritos en los cuadernos de demostración, que implican la generación de una imagen estructural para cada modalidad de imagen. De forma predeterminada, esto implica generar una proyección máxima de la pila z de dos fotones y una imagen media de la grabación de un fotón. Alternativamente, use una imagen media de la grabación de dos fotones.
- Cuando se le solicite, el paso de banda espacial filtra las imágenes para visualizar mejor los puntos de referencia y reorientarlos para que coincidan.
- Seleccione los puntos de referencia coincidentes en las dos imágenes (Figura 6A).
- Utilícelos para calcular la deformación necesaria para alinear las dos imágenes. En general, de 3 a 5 puntos de referencia deberían ser suficientes.
- El algoritmo calcula la deformación en función de una combinación de puntos de referencia y similitud de imágenes. Optimice el peso relativo dado a los dos factores hasta que se logren resultados satisfactorios.
- Deforme el mapa de celdas adquirido en una sesión de un fotón para generar un nuevo mapa de celdas que esté alineado con los datos de dos fotones.
- A continuación, importe este mapa de celdas deformadas en el software de procesamiento 1P para generar una imagen con la imagen de proyección máxima de la película de dos fotones en el fondo.
- Exporte esta imagen para fines de registro.
- En el software de programación, alinee las dos imágenes generadas hasta el momento (mapa de celdas de dos fotones sobre la imagen de proyección máxima de dos fotones) y deforme el mapa de celdas de un fotón sobre la imagen de proyección máxima de dos fotones (Figura 6B, C).
- Para ello, para este estudio, se utiliza una aplicación estimadora de registro que permite al usuario comparar los resultados de diferentes técnicas de registro. Dado que las dos imágenes tienen el mismo fondo, la técnica de correlación de fases, con registro rígido, fue suficiente.
- Una vez completado el registro, escanee la imagen registrada ahora para ver si hay ROI superpuestos. Estos son ROI que están activos en ambas sesiones de grabación, que podrían usarse en un análisis posterior.

Figura 6: Registro de celdas multimodalidad mediante el flujo de trabajo MIRA. (A) Imagen representativa del flujo de trabajo de alineación de celdas. La imagen media de los datos de un fotón se muestra a la izquierda, y la de los datos de dos fotones se muestra a la derecha. Los puntos de referencia coincidentes de ambas imágenes se seleccionan y etiquetan en el software mediante un esquema de color aleatorio (círculos rojos). (B) Las imágenes alineadas por ejemplo que muestran los dos conjuntos de células identificados, un fotón (púrpura) y dos fotones (verde), se superponen a la imagen media de los datos de dos fotones. (C) Imagen de la región marcada con el cuadro blanco en (B), las celdas alineadas se representan aquí como contornos verdes y morados superpuestos. En todos los paneles, la barra de escala representa 200 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Resultados
Se ha demostrado el método de realizar imágenes crónicas de calcio in vivo multicapa de la misma población neuronal durante un período de varias semanas, utilizando modalidades de imágenes de uno y dos fotones, en condiciones de movimiento libre y fijación de la cabeza. Aquí, se ha demostrado la capacidad de identificar poblaciones neuronales coincidentes bajo imágenes de un fotón mientras el animal exploraba una arena abierta en la oscuridad (Figura 7A). Se extrajeron tra...
Discusión
Aquí, hemos demostrado la capacidad de observar y comparar directamente neuronas en condiciones de cabeza fija y movimiento libre en las mismas poblaciones neuronales. Si bien demostramos la aplicación en la corteza visual, este protocolo se puede adaptar a una multitud de otras áreas cerebrales, tanto áreas corticales como núcleos profundos 24,25,26,27,28, así como a otras configuraciones de adquisición de datos y comportamiento
Divulgaciones
Los autores declaran que no hay conflicto de intereses ni intereses contrapuestos.
Agradecimientos
Agradecemos a la Sra. Charu Reddy y al Profesor Matteo Carandini (Cortex Lab) por sus consejos sobre el protocolo quirúrgico y el intercambio de cepas de ratones transgénicos. Agradecemos al Dr. Norbert Hogrefe (Inscopix) por su orientación y asistencia durante el desarrollo de la cirugía. Agradecemos a la Sra. Andreea Aldea (Sun Lab) por su ayuda con la configuración quirúrgica y el procesamiento de datos. Este trabajo fue apoyado por la organización benéfica Moorfields Eye.
Materiales
| Name | Company | Catalog Number | Comments |
| 0.9% Sodium Chloride solution for infusion (Vetivex 11) 250ml | Dechra | 20091607 | Saline for hydration and drug reconsitution |
| 18004-1 Trephine 1.8mm diameter bur | FST | 18004-18 | Drill bit |
| 1ml syringe | Terumo | MDSS01SE | 1ml syringe |
| 23G x 5/8 inch 6% LUER needle | Terumo | NN-2316R | 23G needle |
| 71000 Automated stereotaxic apparatus w/ built-in software | RWD | - | RWD |
| Absorbable Haemostatic Gelatin Sponge (10x10x10mm) | Surgispon | SSP-101010 | gel-foam |
| Alcohol pads 70% isopropyl alcohol | Braun | 9160612 | Alcohol pads |
| Aluminium foil | Any retailer | - | Foil to cover eyes during surgery |
| Articifical Cerebrospinal Fluid | Tocris Bioscience a Bio-Techne Brand | 3525/25ML | ACSF |
| Automated microinjection pump | WPI | 8091 | |
| Betadine solution (10% iodinated Povidone) 500ml | Videne/Ecolab | 3030440 | Betadine |
| Bruker Ultime 2Pplus (customised) | Bruker | - | Two-photon imaging system |
| Cardiff Aldasorber | Vet-Tech | AN006 | Anaesthesia absorber |
| CFI S Plan Fluor ELWD ADM 20XC | Nikon | MRH48230 | 20x objective lens |
| Compact Anaesthesia system - single gas - isoflurane K/F, with oxygen concentrator model: ZY-5AC and scavenging unit | Vet-Tech | AN001 | Compact anaesthesia system |
| Contec Prochlor | Aston Pharma | AP2111L1 | Disinfectant (hypochlorous acid) |
| Dexamethasone Sodium Phosphate Injection, USP, 4mg/ml, NDC: 0641-6145-25 | Hikma | Covetrus:70789 | Dexamethasone |
| Dissecting Knife, cutting edge 4mm, thickness 0.5mm, stainless steel | Fine Science Tools | 10055-12 | Knife for incisino of cortex |
| Dual-Sided, Non-Puncture Mouse & Neonatal Rat Ear Bars | Stoelting | 51649 | Ear bar |
| Dummy microscope | Inscopix | Dummy microscope | To help with implantation |
| Ethanol (100%) | VWR | 40-1712-25 | Used to make 70% ethanol |
| Fisherbrand Nitrile Indigo Disposable Gloves PPE Cat III | FischerScientific | 17182182 | Gloves |
| Homeothermic Monitor 50-7222-F | Harvard Apparatus | 50-7222-F | Homeothermic monitoring system/heating pad |
| Image processing software | ImageJ | - | Image processing software |
| Inscopix Data Processing Software (IDPS) | Inscopix | - | One-photon calcium imaging processing software |
| Insight Duals-232, S/N 2043 | InSight | Insight Spectra X3 | Two-photon imaging laser |
| IsoFlo 250ml 100% w/w inhalation | Zoetis | WM 42058/4195 | Isoflurane |
| Kwik-Sil Low Toxicity Silicone Adhesive | World Precision Intruments (WPI) | KWIK-SIL | Silicone adhesive |
| MICROMOT mains adapter NG 2/S, w/ Drill unit 60/E | PROXXON | NO 28 515 | Handheld drill |
| nVoke Integrated Imaging and Optogenetics System package | Inscopix | - | One-photon Imaging system and software |
| ProView Implant Kit | Inscopix | ProView Implant Kit | Dummy microscope, stereotaxic arm and attachment |
| ProView Prism Probe | Inscopix | 1050-002203 | Microprism lens |
| Rimadyl (50mg/ml) | Zoetis | VM 42058/4123 | Carprofen |
| Stereotaxis Microscope on Articulated arm with table clamp | WPI | PZMTIII-AAC | Microscope |
| Super-Bond Universal kit, SUN Medical | Prestige-Dental | K058E | Adhesive cement |
| Two-photon calcium image software | Suite2P | - | Two-photon calcium imaging processing software |
| Vapouriser | Vet-Tech | - | Isoflurane vapouriser |
| Xailin Lubricating Eye Ointment 5g | Xailin-Night | MLG/28/1551 | Ophthalmic ointment |
Referencias
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Svoboda, K., Yasuda, R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 50 (6), 823-839 (2006).
- Dombeck, D. A., Khabbaz, A. N., Collman, F., Adelman, T. L., Tank, D. W. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 56 (1), 43-57 (2007).
- Vaziri, A., Emiliani, V. Reshaping the optical dimension in optogenetics. Curr Opin Neurobiol. 22 (1), 128-137 (2012).
- Holtmaat, A., et al. high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 4 (8), 1128-1144 (2009).
- Sun, Y. J., Sebastian Espinosa, J., Hoseini, M. S., Stryker, M. P. Experience-dependent structural plasticity at pre- and postsynaptic sites of layer 2/3 cells in developing visual cortex. Proc Natl Acad Sci U S A. 116 (43), 21812-21820 (2019).
- Andermann, M. L., Kerlin, A. M., Reid, R. C. Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci. 4, 3 (2010).
- Sofroniew, N. J., Flickinger, D., King, J., Svoboda, K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. Elife. 5, 14472 (2016).
- Puścian, A., Benisty, H., Higley, M. J. NMDAR-dependent emergence of behavioral representation in primary visual cortex. Cell Rep. 32 (4), 107970 (2020).
- Trachtenberg, J. T., et al. Long-term in vivo. imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 420 (6917), 788-794 (2002).
- Seaton, G., et al. Dual-component structural plasticity mediated by αCaMKII autophosphorylation on basal dendrites of cortical layer 2/3 neurones. J Neurosci. 40 (11), 2228-2245 (2020).
- Helmchen, F., Denk, W. Deep tissue two-photon microscopy. Nat Methods. 2 (12), 932-940 (2005).
- Andermann, M. L., et al. Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron. 80 (4), 900-913 (2013).
- Chia, T. H., Levene, M. J. Microprisms for in vivo multilayer cortical imaging. J Neurophysiol. 102 (2), 1310-1314 (2009).
- Low, R. J., Gu, Y., Tank, D. W. Cellular resolution optical access to brain regions in fissures: Imaging medial prefrontal cortex and grid cells in entorhinal cortex. Proc Natl Acad Sci U S A. 111 (52), 18739-18744 (2014).
- Buxhoeveden, D. P., Casanova, M. F. The minicolumn hypothesis in neuroscience. Brain. 125, 935-951 (2002).
- Chen, S., et al. Miniature fluorescence microscopy for imaging brain activity in freely-behaving animals. Neurosci Bull. 36 (10), 1182-1190 (2020).
- Gulati, S., Cao, V. Y., Otte, S. Multi-layer cortical Ca2+ imaging in freely moving mice with prism probes and miniaturized fluorescence microscopy. J Vis Exp. (124), e55579 (2017).
- Resendez, S. L., et al. Visualization of cortical, subcortical, and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat Protoc. 11 (3), 566-597 (2016).
- Guo, Z. V., et al. Procedures for behavioral experiments in head-fixed mice. PLoS One. 9 (2), 88678 (2014).
- Wekselblatt, J. B., Flister, E. D., Piscopo, D. M., Niell, C. M. Large-scale imaging of cortical dynamics during sensory perception and behavior. J Neurophysiol. 115 (6), 2852-2866 (2016).
- Pnevmatikakis, E. A., et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron. 89 (2), 285-299 (2016).
- Zhou, P., et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife. 7, 28728 (2018).
- Beckmann, L., et al. Longitudinal deep-brain imaging in mouse using visible-light optical coherence tomography through chronic microprism cranial window. Biomed Opt Express. 10 (10), 5235-5250 (2019).
- Wenzel, M., Hamm, J. P., Peterka, D. S., Yuste, R. Reliable and elastic propagation of cortical seizures in. Cell Rep. 19 (13), 2681-2693 (2017).
- Heys, J. G., Rangarajan, K. V., Dombeck, D. A. The functional micro-organization of grid cells revealed by cellular-resolution imaging. Neuron. 84 (5), 1079-1090 (2014).
- Barson, D., Hamodi, A. S. Simultaneous mesoscopic and two-photon imaging of neuronal activity in cortical circuits. Nat Methods. 17 (1), 107-113 (2020).
- Paquelet, G. E., et al. Single-cell activity and network properties of dorsal raphe nucleus serotonin neurons during emotionally salient behaviors. Neuron. 110 (16), 2664-2679 (2022).
- Yang, Q., et al. Transparent microelectrode arrays integrated with microprisms for electrophysiology and simultaneous two-photon imaging across cortical layers. bioRxiv. , (2022).
- Priestley, J. B., Bowler, J. C., Rolotti, S. V., Fusi, S., Losonczy, A. Signatures of rapid plasticity in hippocampal CA1 representations during novel experiences. Neuron. 110 (12), 1978-1992 (2022).
- Zong, W., et al. Miniature two-photon microscopy for enlarged field-of-view, multi-plane and long-term brain imaging. Nat Methods. 18 (1), 46-49 (2021).
- Engelbrecht, C. J., et al. Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express. 16 (8), 5556-5564 (2008).
- Suzuki, M., Aru, J., Larkum, M. E. Double-μ Periscope, a tool for multilayer optical recordings, optogenetic stimulations or both. Elife. 10, 72894 (2021).
- Stibůrek, M., et al. 110 μm thin endo-microscope for deep-brain in vivo observations of neuronal connectivity, activity and blood flow dynamics. Nat Commun. 14 (1), 1897 (2023).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados