
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Imaging a lungo termine di popolazioni neurali identificate utilizzando microprismi in animali che si muovono liberamente e fissano la testa
In questo articolo
Riepilogo
Se integrata con una piastra di testa e un design ottico compatibile con microscopi a singolo e due fotoni, la lente del microprisma presenta un vantaggio significativo nella misurazione delle risposte neurali in una colonna verticale in diverse condizioni, inclusi esperimenti ben controllati in stati di fissazione della testa o compiti comportamentali naturali in animali che si muovono liberamente.
Abstract
Con il progresso della microscopia multi-fotone e delle tecnologie molecolari, l'imaging a fluorescenza sta rapidamente crescendo fino a diventare un potente approccio per studiare la struttura, la funzione e la plasticità dei tessuti cerebrali viventi. Rispetto all'elettrofisiologia convenzionale, la microscopia a fluorescenza è in grado di catturare l'attività neurale e la morfologia delle cellule, consentendo registrazioni a lungo termine delle popolazioni di neuroni identificate a risoluzione singola o subcellulare. Tuttavia, l'imaging ad alta risoluzione richiede in genere una configurazione stabile e fissata sulla testa che limita il movimento dell'animale, e la preparazione di una superficie piana di vetro trasparente consente la visualizzazione dei neuroni su uno o più piani orizzontali, ma è limitata nello studio dei processi verticali che attraversano diverse profondità. Qui, descriviamo una procedura per combinare una fissazione della piastra di testa e un microprisma che fornisce immagini multistrato e multimodali. Questa preparazione chirurgica non solo dà accesso all'intera colonna della corteccia visiva del topo, ma consente l'imaging a due fotoni in una posizione fissa sulla testa e l'imaging a un fotone in un paradigma che si muove liberamente. Utilizzando questo approccio, è possibile campionare popolazioni cellulari identificate in diversi strati corticali, registrare le loro risposte in stati di testa fissa e in movimento libero e monitorare i cambiamenti a lungo termine nel corso dei mesi. Pertanto, questo metodo fornisce un saggio completo dei microcircuiti, consentendo il confronto diretto delle attività neurali evocate da stimoli ben controllati e secondo un paradigma comportamentale naturale.
Introduzione
L'avvento dell'imaging fluorescente a due fotoni in vivo 1,2, che combina le nuove tecnologie nei sistemi ottici e gli indicatori di fluorescenza geneticamente modificati, è emerso come una potente tecnica nelle neuroscienze per studiare l'intricata struttura, funzione e plasticità nel cervello vivente 3,4. In particolare, questa modalità di imaging offre un vantaggio senza precedenti rispetto all'elettrofisiologia tradizionale, catturando sia la morfologia che le attività dinamiche dei neuroni, facilitando così il monitoraggio a lungo termine dei neuroni identificati 5,6,7,8.
Nonostante i suoi notevoli punti di forza, l'applicazione dell'imaging a fluorescenza ad alta risoluzione richiede spesso una configurazione statica e fissa sulla testa che limita la mobilità dell'animale 9,10,11. Inoltre, l'uso di una superficie di vetro trasparente per la visualizzazione dei neuroni restringe le osservazioni a uno o più piani orizzontali, limitando l'esplorazione della dinamica dei processi verticali che si estendono a diverse profondità corticali12.
Affrontando queste limitazioni, il presente studio delinea una procedura chirurgica innovativa che integra la fissazione della placca testale, il microprisma e il miniscopio per creare una modalità di imaging con capacità multistrato e multimodale. Il microprisma consente l'osservazione dell'elaborazione verticale lungo la colonna corticale 13,14,15,16, che è fondamentale per capire come l'informazione viene elaborata e trasformata mentre si muove attraverso i diversi strati della corteccia e come l'elaborazione verticale viene alterata durante i cambiamenti plastici. Inoltre, consente l'imaging delle stesse popolazioni neurali in un paradigma fissato alla testa e in un ambiente in movimento libero, comprendendo le versatili impostazioni sperimentali 17,18,19: ad esempio, la fissazione della testa è spesso richiesta per paradigmi ben controllati come la valutazione della percezione sensoriale e le registrazioni stabili sotto il paradigma a 2 fotoni, mentre il movimento libero offre un ambiente più naturale e flessibile per gli studi comportamentali. Pertanto, la capacità di condurre un confronto diretto in entrambe le modalità è fondamentale per approfondire la nostra comprensione dei microcircuiti che consentono risposte flessibili e funzionali.
In sostanza, l'integrazione della fissazione della placca testa, del microprisma e del miniscopio nell'imaging a fluorescenza offre una piattaforma promettente per sondare le complessità della struttura e della funzionalità del cervello. I ricercatori possono campionare le popolazioni cellulari identificate a varie profondità che coprono tutti gli strati corticali, confrontare direttamente le loro risposte in paradigmi sia ben controllati che naturali e monitorare le loro alterazioni a lungo termine per mesi20. Questo approccio offre preziose informazioni su come queste popolazioni neurali interagiscono e cambiano nel tempo in diverse condizioni sperimentali, fornendo una finestra sulla natura dinamica dei circuiti neurali.
Protocollo
Tutti gli esperimenti sono stati condotti secondo l'UK Animals (Scientific Procedures) Act 1986 in base a licenze personali e di progetto approvate e rilasciate dal Ministero degli Interni del Regno Unito a seguito di un'appropriata revisione etica. Linee transgeniche adulte CaMKII-TTA; GCaMP6S-TRE21 sono stati allevati e la loro progenie è stata utilizzata nell'esperimento. Per la sicurezza degli sperimentatori e il mantenimento delle condizioni di sterilità, tutte le procedure sono state eseguite in condizioni asettiche e con dispositivi di protezione individuale completi.
1. Preparazione pre-operatoria
- Per ridurre al minimo l'edema, somministrare Desametasone (0,2 mg/kg) per via sottocutanea, 12-24 ore prima dell'intervento.
- Sterilizzare tutti gli strumenti chirurgici in autoclave e sterilizzare l'area chirurgica con acido ipocloroso stabilizzato con acqua distillata ed etanolo al 70% prima dell'intervento chirurgico. Assicurarsi che tutte le apparecchiature chirurgiche siano accese.
- Anestetizzare l'animale (24 settimane di età, maschio del peso di 31 g) utilizzando isoflurano con una dose di induzione del 5%, che viene ridotta all'1%-2% una volta che il topo è sul telaio stereotassico, con O2 mantenuto tra 1-2 L/min. Iniettare FANS (Carprofene, 2,5 mg/kg) per via sottocutanea.
- Verificare l'assenza del riflesso di pizzicamento delle dita dei piedi per valutare la profondità dell'anestesia (aumentare la concentrazione di isoflurano con incrementi dello 0,5% se si vede il riflesso).
- Radere la testa dell'animale, usando un rifinitore, da dietro le orecchie fino a poco sopra gli occhi. Pulisci quest'area con una salvietta imbevuta di alcol e una soluzione di iodio povidone, assicurandoti di evitare il contatto con gli occhi dell'animale.
- Montare l'animale sul termoforo omeotermico e sul telaio stereotassico dotato di barre per le orecchie e i denti e fissare la testa. Assicurarsi che la testina sia stabile, poiché ciò è fondamentale per il successo della seguente procedura (Figura 1).
- Applicare un unguento oftalmico sugli occhi dell'animale per evitare che si secchino durante l'intervento chirurgico e coprirli con un foglio per proteggerli dalla luce. Coprire l'animale con una copertura chirurgica sterile.

Figura 1: Preparazione pre-operatoria. Il mouse è posizionato sul telaio stereotassico, fissato da un nasello e da barre auricolari. Il mouse è posizionato su un pad riscaldato a temperatura regolata. Gli occhi hanno un unguento oftalmico su di loro e sono coperti da un foglio di alluminio. La testa è rasata e il cranio è esposto. Una copertura sterile viene posta sopra l'animale. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
2. Craniotomia
- Usando le forbici chirurgiche, incidi la pelle lungo la linea mediana dell'area rasata della testa per esporre il cranio.
- Pulire il cranio utilizzando un batuffolo di cotone sterile e perossido di idrogeno diluito (3% p/v di 35% H2O2 in 97% dH2O) per 1-3 secondi per rimuovere il tessuto connettivo (Figura 2A). Asciugare il cranio utilizzando una goccia di etanolo al 70% e un nuovo batuffolo di cotone sterile.
- Allineare il cranio anteriormente/posteriormente (AP) e medialmente/lateralmente (ML) per garantire un sito di impianto accurato. Per fare ciò, misurare la profondità dorso-ventrale (DV) del cranio sia in bregma che in lambda e assicurarsi che la differenza tra i due sia di <0,03 mm. Per l'allineamento mediolaterale, misurare i punti equidistanti su entrambe le ossa parietali dalla linea mediana e assicurarsi che la differenza DV sia di <0,03 mm.
- Usando il bregma come origine, trova e segna l'area corticale desiderata; in questo caso, si tratta di corteccia visiva primaria monoculare (V1), AP: -3,5 mm, ML: -2,5 mm.
- Utilizzare una punta da trapano a trefina (diametro 1,8 mm) e un trapano dentale (velocità di 10.000 giri/min) per esporre la corteccia, assicurandosi che il segno per l'area corticale desiderata (monoculare V1) si trovi all'interno del terzo inferiore della finestra della punta del trapano.
- Assicurarsi che l'angolo della punta del trapano sia perpendicolare alla curvatura del cranio. Ciò garantirà una craniotomia uniforme e preverrà danni alla dura o alla corteccia.
- Forare fino a quando non si verifica una diminuzione della resistenza e quindi fermarsi (Figura 2B). Rimuovere con cautela il frammento osseo staccato utilizzando la punta di un ago da 23G (Figura 2C).
- Pulire la corteccia esposta con schiuma chirurgica imbevuta di liquido cerebrospinale artificiale freddo (ACSF) per rimuovere eventuali detriti e fermare eventuali emorragie che potrebbero verificarsi.
- Mantieni sempre idratata la corteccia esposta, utilizzando ACSF freddo, durante l'intervento.

Figura 2: Craniotomia. (A) Viene mostrata l'incisione cutanea tra bregma e lambda. Il tessuto connettivo è stato rimosso dalla superficie esposta. (B) Craniotomia mediante trapano a trefina prima della rimozione del frammento osseo. (C) Craniotomia dopo la rimozione di un frammento osseo, che mostra la dura e la corteccia intatte (la barra della scala rappresenta 0,5 mm). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
3. Incisione pretagliata
NOTA: Per essere presi in considerazione quando si esegue l'incisione pretagliata, l'incisione e l'impianto del microprisma dovranno essere anteriori alla regione di interesse (ROI) dell'imaging. Questo per consentire un campo visivo completo e preciso. Nell'ambito di questo protocollo, l'incisione verrà eseguita lungo l'asse mediolaterale e il microprisma sarà orientato verso la parte posteriore (Figura 3B).
- Per facilitare l'inserimento e alleviare la pressione nella corteccia durante l'inserimento del microprisma, praticare un'incisione.
- Fissare il coltello chirurgico al supporto del braccio stereotassico e orientare la lama o il braccio stereotassico in modo che tagli lungo l'asse ML.
- Spostare il coltello sulla coordinata AP desiderata (AP: -3.4 mm); il prisma deve essere di fronte alla ROI, quindi eseguire l'incisione 100 μm anteriormente alla coordinata AP della ROI di imaging (-3,5 mm).
- Ora spostando il coltello sul bordo mediale della craniotomia, dove incontra il cranio, abbassare lentamente il coltello fino a raggiungere l'osso e poi fermarsi. Poiché lo spessore dell'osso è di 200 μm, incorporare questo valore nella profondità di inserzione totale (vedere il passaggio 5.1 di calcolo).
- L'imaging ottimale è al centro del prisma, cioè 500 μm; pertanto, assicurarsi che questa profondità sia allineata con la profondità della colonna corticale (ROI DV: - 0,35 mm).
- Incorporando lo spessore del cranio nel calcolo della profondità, usa l'equazione seguente, che determina quanto deve essere profonda l'incisione pretagliata dalla superficie del cranio. Per questo protocollo, la profondità di impianto viene calcolata come:
Spessore osseo (200 μm) + ROI di imaging (ad es. 350 μm) + profondità residua del microprisma (500 μm) = 1.050 μm
- Incorporando lo spessore del cranio nel calcolo della profondità, usa l'equazione seguente, che determina quanto deve essere profonda l'incisione pretagliata dalla superficie del cranio. Per questo protocollo, la profondità di impianto viene calcolata come:
- Assicurarsi che la lunghezza dell'incisione sia superiore a 1 mm ma non eccessiva; pertanto, una distanza di 1,2 mm è l'ideale, con la coordinata ML monoculare che si trova nel mezzo di questa distanza.
- Quando si è pronti per eseguire l'incisione, rimuovere l'ACSF in eccesso in modo che la visione non sia oscurata (Figura 3A).
- Spostare il coltello dal bordo mediale della craniotomia alla coordinata mediale iniziale dell'incisione. Abbassare lentamente (10 μm/s) il coltello nella corteccia.
- Una volta che la dura è stata perforata e il coltello è entrato nella corteccia, applicare una goccia di ACSF freddo sulla corteccia per mantenere il tessuto lubrificato e idratato durante l'incisione.
- Una volta raggiunta la profondità finale, iniziare a muovere il coltello lungo l'asse ML (ad una velocità di 10 μm/s).
- Continua a osservare il tessuto circostante mentre viene praticata l'incisione. Se il tessuto si trascina con il coltello, muovi il coltello su e giù alcune volte per assicurarti che il tessuto venga tagliato, quindi continua lateralmente, ricordandoti di rimettere il coltello alla sua profondità finale.
- Al termine, solleva lentamente il coltello. Se il sangue emerge durante l'incisione, utilizzare questo tempo per pulire il sito dell'incisione con schiuma chirurgica imbevuta di ACSF per diluire il sangue e spingere fuori il sangue all'interno dell'incisione. Ricordarsi di lasciare una schiuma chirurgica fresca e satura sulla corteccia esposta fino al momento di inserire il microprisma.

Figura 3: Impianto di microprisma. (A) Incisione pretagliata. (B) Schema della lente microprismatica integrata che mostra la sua posizione all'interno della corteccia (C) Lente microprismatica integrata con l'orientamento corretto per pretagliare l'incisione prima dell'inserimento nella corteccia (la barra della scala rappresenta 0,5 mm). (D) Esempio di accumulo di cemento attorno alla lente integrata per fissarne l'attacco al cranio. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
4. Inserimento del microprisma e impianto della placca di testa
- La lente microprismatica è composta da una lente con indice di gradiente attaccata a un prisma all'estremità distale della lente, che è integrata in una piastra di base. Fissare il microprisma al kit dell'impianto.
- Assicurarsi che il lato imaging del prisma sia opposto alla vite della piastra di base. Per facilitare l'inserimento e il posizionamento del microprisma, fissarlo al telaio stereotassico e orientare il prisma in modo che sia allineato con l'incisione (Figura 3C).
- Abbassare lentamente il microprisma nel sito di incisione (10 μm/s). Ricordarsi di rimuovere l'ACSF quando si inserisce inizialmente il prisma, ma una volta nella corteccia, sciacquare con ACSF freddo per lubrificare l'inserimento.
- La corteccia dovrebbe rimanere stabile mentre il prisma viene abbassato nell'incisione; in caso contrario, aggiungere ulteriore ACSF e agitare la corteccia muovendo il prisma su e giù per allentare la corteccia dal prisma.
- Una volta raggiunta la profondità finale, asciugare la superficie corticale esposta utilizzando tessuto sterile, facendo attenzione a non toccare il prisma.
- Coprire l'area corticale esposta che circonda il prisma, così come la lente, con uno strato protettivo di adesivo siliconico, riducendo al minimo l'eccesso di adesivo sul cranio circostante e sul cannocchiale fittizio.
- Una volta indurito (5-10 min), fissare la placca al cranio per stabilizzare la testa durante l'imaging con fissaggio alla testa.
- Assicurarsi che la placca sia sufficientemente posteriore da non interferire con il posizionamento dell'impianto e da consentire un'adeguata applicazione di cemento per fissare correttamente l'impianto.
- Assicurarsi che la linea mediana della placca della testina si trovi leggermente a destra della lente impiantata per garantire che entrambi i lati della piastra della testa possano essere fissati al palco della testa durante l'esecuzione di esperimenti con fissaggio alla testa
- Applicare del cemento adesivo sulla placca e sul cranio.
- Preparare il cemento dentale adesivo mescolando 1 misurino di polvere di cemento opaco con 4 gocce di mezzo di miscelazione e applicando una goccia di catalizzatore.
- Posiziona il cemento sia sulla corazza che sul cranio e tieni la placca in posizione fino a quando non si è indurita, assicurandoti che sia parallela alle barre auricolari (attraverso l'esame visivo, ispezionare sia dall'alto che da dietro la testa dell'animale).
- Applicare del cemento adesivo per coprire il resto del cranio e del tessuto esposti, incorporando il microprisma (fino alla base della piastra di base) e la placca della testa.
- Non mettere cemento sulla piastra di base, sul microscopio fittizio o su uno qualsiasi dei suoi componenti. Continuare ad applicare il cemento adesivo fino a quando il microprisma e la piastra di testa non sono coperti e sono stabili (Figura 3D).
- Quando il cemento si è indurito, staccare il microscopio fittizio muovendo lentamente il braccio stereotassico verso l'alto stabilizzando il microprisma con una pinza (sono collegati tramite magneti, quindi si potrebbe avvertire una certa resistenza durante la separazione).
- Inserire la copertura protettiva sull'obiettivo e serrare la vite per fissarla in posizione.
- Rimuovere l'animale dalla cornice stereotassica, lasciarlo riprendersi in una scatola di recupero calda e somministrare soluzione fisiologica sterile riscaldata allo 0,9% per via sottocutanea (3% del peso corporeo).
- Una volta che l'animale è sveglio e in movimento, rimettilo in una gabbia singola pulita. Monitorare l'animale e somministrare ulteriori analgesia post-operatorie secondo la politica dell'istituzione locale sull'analgesia.
- Attendere 4 settimane dopo l'intervento, l'animale dovrebbe essere pronto per l'imaging.
5. Imaging del calcio a un fotone degli strati corticali in topi che si muovono liberamente
NOTA: È essenziale utilizzare ogni volta le immagini acquisite dalla sessione di imaging originale per garantire un'acquisizione accurata del piano di imaging previsto. Questi punti di riferimento identificati, insieme ai neuroni, svolgono un ruolo fondamentale nel processo di allineamento descritto in dettaglio nella fase 9 del protocollo. Quando si acquisiscono dati a un fotone, il miniscopio è sia il sistema di imaging che la sorgente laser. L'eccitazione utilizza LED con un intervallo di potenza di 0-2 mW/mm2 sulla superficie anteriore dell'obiettivo. Il laser utilizza una lunghezza d'onda di eccitazione di 455 ± 8 nm (luce blu) per la segnalazione GCaMP. Il cursore della messa a fuoco dell'obiettivo può essere utilizzato per regolare la messa a fuoco (asse Z), rappresentata sull'interfaccia come 0-1000, dove 0 rappresenta una distanza di lavoro di 0 μm e 1000 rappresenta la distanza di lavoro massima di 300 μm.
- Prima di acquisire i dati, lasciare che l'animale si acclimati alla stanza e all'arena aperta per 1 ora prima della sessione di registrazione.
- Prima dell'imaging, disinfettare e pulire il tutto con disinfettanti appropriati (es. acido ipocloroso stabilizzato con acqua distillata ed etanolo al 70%).
- Configurare il DAQ box collegandolo a un computer e avviando il software di acquisizione dati. Stabilire una connessione diretta tramite un cavo ethernet per ridurre al minimo i frame caduti; Tuttavia, la modalità di connessione wireless potrebbe essere sufficiente a seconda della potenza della connessione wireless.
- Attacca il miniscopio alla piastra di base dell'animale sotto una collottola delicata.
- Innanzitutto, rimuovere il coperchio protettivo dalla piastra di base svitando la vite di fermo. Tenere il coperchio per l'apertura con una pinza. Quindi, fissare il miniscopio alla piastra di base, dove si trova il coperchio.
- Controllare l'orientamento del miniscopio rispetto alla piastra di base prima di installarlo in modo che il lato con la marcatura della vite sia rivolto verso la vite.
- Una volta fissato il miniscopio, serrare la vite di fermo per stabilizzarlo. Far avanzare la vite di fermo solo fino a quando non si avverte una certa resistenza. Un serraggio eccessivo della vite di fermo potrebbe danneggiare il miniscopio e dovrebbe quindi essere evitato.
- Collegare il miniscopio al DAQ box e preparare il software per la registrazione.
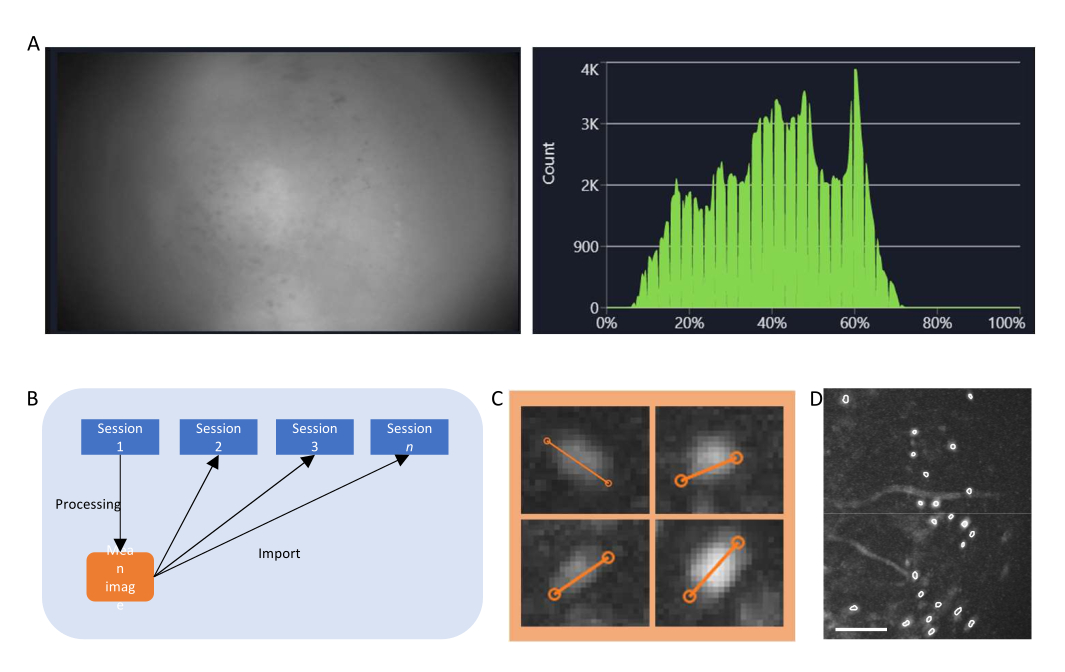
- Nel software di acquisizione dati, attivare il flusso per il miniscopio e regolare i parametri di registrazione (frame rate di imaging, guadagno, potenza LED, valore efocus) per ottenere un campo visivo chiaro (Figura 4A).
- Accendere la finestra dell'istogramma e regolare il guadagno e la potenza del LED in modo che l'intensità registrata sia compresa tra il 35% e il 70%.
- Se si esegue uno studio longitudinale, fare riferimento alle registrazioni o alle istantanee scattate in una sessione precedente e regolare il valore di messa a fuoco in modo che lo stesso piano di imaging sia chiaramente visibile.
- Avviare l'esperimento e l'acquisizione dei dati.
- Dopo il completamento dell'esperimento, rimuovere l'animale dall'apparato comportamentale.
- Sotto una collottola delicata, allentare la vite di fermo e rimuovere il miniscopio dalla piastra di base dell'animale.
- Riposizionare il coperchio di protezione sulla piastra di base e stabilizzarlo con la vite di fermo.
- Riportare l'animale nella sua gabbia di casa (procedere al passaggio 7 se si desidera elaborare i dati a un fotone).

Figura 4: Acquisizione ed elaborazione dei dati con software. (A) Un'immagine che mostra il flusso in tempo reale dal miniscopio. Si consiglia di regolare il valore di messa a fuoco dell'obiettivo, in modo che nella finestra di streaming venga visualizzata una visione chiara, insieme al guadagno e alla potenza del laser di imaging (B) Grafico schematico che illustra il flusso di lavoro di allineamento consigliato per le sessioni registrate in diversi punti temporali. Si consiglia di generare un'immagine media fin dalla prima sessione, seguendo le istruzioni per il software di elaborazione dati. Questa immagine deve essere utilizzata come immagine di riferimento durante la correzione del movimento per le sessioni successive. (C) Esempi di quattro celle della stessa immagine ΔF/F proiettata al massimo. Una linea arancione viene tracciata su ogni cella per misurarne il diametro in pixel, la cui media viene presa come argomento di input per l'algoritmo di identificazione delle celle (in alto a sinistra: 13, in alto a destra: 11, in basso a sinistra: 12, in basso a destra: 13). (D) Output dell'algoritmo di identificazione delle celle dopo la cura manuale (immagine ritagliata). I contorni bianchi rappresentano le celle identificate (la barra della scala rappresenta 100 μm). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
6. Imaging del calcio a due fotoni degli strati corticali in topi con fissaggio alla testa
NOTA: Per la microscopia a scansione laser a due fotoni, la sorgente luminosa è un laser ultraveloce sintonizzabile con una lunghezza d'onda di eccitazione di 920 nm. La potenza di eccitazione, misurata all'obiettivo, era in genere compresa tra 100 e 150 mW e regolata in ogni sessione per ottenere livelli simili di fluorescenza. La luce di emissione è stata filtrata da un filtro di emissione (525/70 nm) e misurata da un tubo fotomoltiplicatore indipendente (PMT), denominato canale verde. Le immagini sono state acquisite con un obiettivo ad immersione in aria 20x (NA = 0,45, distanza di lavoro 6,9-8,2 mm).
- Prima di acquisire i dati, abituare l'animale all'apparecchio nei giorni precedenti (per far sì che l'animale si senta a proprio agio con l'impostazione comportamentale). Lasciare che l'animale trascorra 15-30 minuti al giorno, per 2-3 giorni, esplorando l'ambiente o fino a quando non mostra un comportamento naturalistico prima di iniziare l'acquisizione dei dati.
- Accendere il sistema di imaging a due fotoni, avviare il software di acquisizione e accendere il laser. Assicurarsi che i laser siano chiusi e che i PMT siano spenti prima di continuare.
- Pulire l'apparecchio di imaging a due fotoni con acido ipocloroso stabilizzato con acqua distillata ed etanolo al 70%.
- Assicurati di regolare l'apparecchio per adattarlo alle dimensioni dell'animale. Fissare delicatamente il mouse e la sua piastra di testa alla configurazione del tavolino di testa e avvitarla in posizione per stabilizzare la testa del mouse.
- Una volta fissato, rimuovere il copriobiettivo (fare riferimento al punto 5.4.1) e allineare l'obiettivo in modo che si trovi sopra la piastra di base.
- Utilizzare i controlli dello stadio di epifluorescenza e XYZ per mettere a fuoco il tessuto corticale.
- Una volta che gli strati corticali sono visibili, cambiare il microscopio per consentire l'imaging a due fotoni (sostituire lo specchio con lo specchio dicroico, chiudere l'otturatore fluorescente e spegnere il laser a epifluorescenza e il monitor). Assicurarsi di spegnere le luci principali in modo da proteggere i PMT durante l'imaging.
- Impostare i parametri per ottimizzare i file immagine acquisiti.
- Utilizzare la modalità di acquisizione risonante per l'imaging del calcio in quanto è in grado di catturare l'attivazione rapida dei segnali GCaMP.
- Regola la potenza del laser, il guadagno PMT, lo zoom e le tabelle di ricerca (LUT) per ottenere un'immagine ottimale e fai riferimento all'immagine a un fotone per assicurarti che venga ripreso il piano focale corretto.
- Iniziare l'imaging degli strati corticali con il sistema a due fotoni con monitoraggio comportamentale sincronizzato e input di stimolo (se applicabile).
- Assicurarsi di salvare questi parametri di acquisizione e le distanze XYZ se si desidera riprodurre immagini identiche nel tempo.
- Per acquisire una pila z degli strati corticali, seguire i passaggi descritti di seguito.
- Trova il piano da cui iniziare lo z-stack, regola i parametri di acquisizione per ottimizzare l'immagine e contrassegnalo come punto di partenza sul software.
- Successivamente, utilizzando il controllo Z, spostati verso il basso nella pila, regolando solo la potenza del laser per mantenere una luminanza costante della pila e contrassegna la fine della pila sul software.
- Passaggio critico: utilizzando l'opzione Gradiente esponenziale relativo nella scheda Gradiente di potenza laser , consente al software di calcolare l'aumento della potenza del laser mentre si muove attraverso lo z-stack. Assicurati di segnare i valori di potenza del laser finale nella tabella fornita dal software per consentirgli di calcolare il gradiente.
- Una volta impostati i parametri dello z-stack, regolare la dimensione del passo (μm).
NOTA: la dimensione del passaggio determinerà il tempo impiegato, il numero di sezioni e la qualità dei dettagli dello stack. Passi di dimensioni più piccole si tradurranno in un tempo di acquisizione più lungo, un aumento del numero di sezioni e un migliore dettaglio rispetto a passi di dimensioni maggiori. Gli Z-stack vengono utilizzati per assistere nella registrazione di immagini a uno e due fotoni, in quanto evidenziano eventuali punti di riferimento o caratteristiche anatomiche.
- Per acquisire una serie temporale (serie T) di cambiamenti di calcio nei neuroni, trovare un piano focale ottimale utilizzando i controlli dello stadio XYZ e regolare la potenza del laser, il guadagno PMT, lo zoom e le LUT.
- Nella scheda Serie T del software di acquisizione, definire i parametri della frequenza di acquisizione in modo che corrispondano ai dati acquisiti utilizzando il sistema di imaging a un fotone.
NOTA: La corrispondenza della frequenza renderà i dati 1P e 2P comparabili ed è più adatta per l'algoritmo di identificazione delle celle utilizzato nelle fasi di elaborazione dei dati. Trigger multipli e altre modalità di acquisizione possono essere incorporati nell'acquisizione della serie T.
- Nella scheda Serie T del software di acquisizione, definire i parametri della frequenza di acquisizione in modo che corrispondano ai dati acquisiti utilizzando il sistema di imaging a un fotone.
- Inizia l'acquisizione della serie T.
7. Elaborazione dei dati di imaging del calcio a un fotone
- Per la registrazione di filmati con un fotone, utilizzare il software di elaborazione dati fornito con il sistema miniscope.
- Innanzitutto, pre-elabora il filmato mediante un downsampling spaziale e temporale. In generale, il downsampling spaziale del filmato di un fattore due ridurrebbe significativamente il tempo di elaborazione senza compromettere gravemente l'accuratezza dell'identificazione delle cellule.
- Impostare il fattore di downsampling temporale in modo che la frequenza dei fotogrammi del filmato venga ridotta a circa 10 Hz, che è più adatta per l'algoritmo di identificazione delle celle utilizzato nei passaggi seguenti.
- Se si acquisiscono più filmati nello stesso giorno di imaging, combinare i filmati in un'unica serie temporale prima della fase di pre-elaborazione per elaborarli insieme.
- Facoltativo: applica un filtro passa-banda spaziale al filmato per rimuovere le frequenze spaziali più basse e più alte, ottenendo un filmato più fluido con un contrasto più elevato.
- Registrare il filmato utilizzando la funzionalità di correzione del movimento del software. In questo modo il filmato viene registrato e vengono corretti gli artefatti da movimento causati dal movimento del miniscopio rispetto alla superficie dell'immagine.
- Passaggio critico: se si esegue uno studio longitudinale, registrare i filmati nello stesso campo visivo, ad esempio l'immagine media del filmato ripreso il primo giorno di imaging (Figura 4B).
- Calcola il ΔF/F del filmato utilizzando la scheda corrispondente e proietta il filmato per generare un'immagine di proiezione massima del filmato ΔF/F. Questa immagine mostrerà le regioni che mostrano cambiamenti nei livelli di fluorescenza, potenzialmente singoli neuroni, e potrebbe essere utilizzata per misurare il diametro medio dei neuroni (Figura 4C).
- In alternativa, misurare il diametro della cellula sul film con correzione del movimento, dove i neuroni mostrano una chiara fluorescenza.
- Identifica le cellule utilizzando gli algoritmi del software.
- Sebbene in questa fase siano disponibili due opzioni (PCA-ICA e CNMF-E), utilizzare CNMF-E per questo studio. Immettere il diametro medio della cella in pixel ed eseguire l'algoritmo per generare un set di celle contenente regioni di interesse (ROI) che dimostrano attività simili a quelle delle celle.
- Selezionare manualmente le ROI che sono cellule (hanno una morfologia simile a quella cellulare e un'attività22,23 e si trovano all'interno del FOV) da quelle che non lo sono e convalidare il set di celle curato (Figura 4D).
- Esporta le tracce di calcio di ogni ROI per ulteriori analisi.
8. Elaborazione dei dati di imaging del calcio a due fotoni
- Per la registrazione di filmati a due fotoni, utilizzare un pacchetto python progettato per elaborare i dati di analisi del calcio a due fotoni.
- Innanzitutto, combina le immagini scattate in una serie T in una pila .tiff come descritto di seguito.
- Nell'interfaccia delle opzioni di esecuzione , regolare i parametri, inclusi il valore tau e la frequenza dei fotogrammi, in modo che corrispondano al GCaMP utilizzato e alla frequenza dei fotogrammi della registrazione.
- Facoltativo: impostare il parametro do_registration su 1 per registrare il filmato. Ciò equivale alla fase di correzione del movimento descritta in precedenza.
- Facoltativo: impostare il parametro anatomical_only su 1 per rilevare le ROI utilizzando le caratteristiche anatomiche oltre alla dinamica della fluorescenza. Ciò richiede l'inserimento del diametro della cella, quindi eseguire le misurazioni utilizzando il software di elaborazione delle immagini. Questa operazione è generalmente consigliata, in quanto genera ROI con forme più naturali (Figura 5A-C).
- Una volta impostati tutti i parametri, eseguire l'algoritmo in modo che esegua tutti i calcoli insieme. Fare riferimento all'interfaccia utente grafica (GUI) per verificare l'avanzamento.
- Al termine, torna all'interfaccia di selezione delle celle per la cura manuale dei risultati di identificazione delle celle.
- Salvate un'immagine della cella curata sopra la proiezione massima del filmato. Questa verrà utilizzata in seguito come immagine di riferimento per la registrazione dei dati di registrazione di un fotone.
- L'algoritmo salva quindi automaticamente i risultati in. npy, a cui si può accedere in seguito con Python. In alternativa, salvare i risultati in altri file formattati per ulteriori analisi in altri software.

Figura 5: Identificazione delle cellule utilizzando un software di elaborazione a due fotoni. (A) Immagine rappresentativa dell'identificazione delle cellule ripresa dal software di elaborazione a due fotoni. Impostando Anatomical_only parametro su 0 ma mantenendo invariati tutti gli altri parametri, nell'area tra le linee tratteggiate sono presenti più celle non celle che interferiscono con la cura manuale delle celle effettive. (B) Esempi di misure di diametro cellulare tratte da (A), utilizzando un software di elaborazione delle immagini (in alto a sinistra; 7,5 pixel, in alto a destra; 9, in basso a sinistra; 6,5, in basso a destra; 7,5). (C) Immagine rappresentativa dell'identificazione cellulare. Quando si imposta Anatomical_only parametro su 1 e si inserisce il diametro medio della cella preso da (B) nell'algoritmo del diametro della cella, non sono presenti celle nell'area tra le linee tratteggiate (le barre della scala rappresentano 200 μm). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
9. Registrazione di set cellulari identificati attraverso le modalità di imaging
- Eseguire la registrazione di cellule identificate da registrazioni a uno e due fotoni con l'algoritmo di registrazione e analisi dell'immagine multimodale (MIRA), disponibile tramite l'interfaccia Python del software di imaging a un fotone.
- Questo algoritmo allinea i dati a un fotone e a due fotoni tramite una registrazione non rigida. Vedere la serie di quaderni dimostrativi online trovati sul sito Web e utilizzati per questo studio.
NOTA: I notebook sono stati scritti in modo che tutta l'elaborazione venga completata nel software 1P e non sono quindi compatibili con il software di elaborazione 2P. Pertanto, segui solo alcuni dei passaggi nei quaderni per questo studio.
- Questo algoritmo allinea i dati a un fotone e a due fotoni tramite una registrazione non rigida. Vedere la serie di quaderni dimostrativi online trovati sul sito Web e utilizzati per questo studio.
- Seguire i passaggi descritti nei notebook dimostrativi, che prevedono la generazione di un'immagine strutturale per ogni modalità di imaging. Per impostazione predefinita, ciò comporta la generazione di una proiezione massima dello stack z a due fotoni e un'immagine media della registrazione a un fotone. In alternativa, utilizzare un'immagine media della registrazione a due fotoni.
- Laddove richiesto, la banda passante spaziale filtra le immagini per visualizzare meglio i punti di riferimento e riorientarli in modo che corrispondano.
- Selezionare i punti di riferimento corrispondenti sulle due immagini (Figura 6A).
- Usateli per calcolare l'alterazione necessaria per allineare le due immagini. In generale, da 3 a 5 punti di riferimento dovrebbero essere sufficienti.
- L'algoritmo calcola la deformazione in base a una combinazione di punti di riferimento e somiglianza dell'immagine. Ottimizzare il peso relativo dato ai due fattori fino a ottenere risultati soddisfacenti.
- Distorcere la mappa cellulare acquisita in una sessione a un fotone per generare una nuova mappa a celle allineata ai dati a due fotoni.
- Quindi importa questa mappa di celle deformate nel software di elaborazione 1P per generare un'immagine con l'immagine di proiezione massima dal filmato a due fotoni sullo sfondo.
- Esporta questa immagine a scopo di registrazione.
- Nel software di programmazione, allineare le due immagini generate finora (mappa delle celle a due fotoni sopra l'immagine di proiezione massima a due fotoni) e deformare la mappa delle celle a un fotone sopra l'immagine di proiezione massima a due fotoni (Figura 6B,C).
- A tale scopo, per questo studio, utilizzare un'applicazione di stima della registrazione che consente all'utente di confrontare i risultati di diverse tecniche di registrazione. Dato che le due immagini hanno lo stesso sfondo, è stata sufficiente la tecnica della correlazione di fase, con registrazione rigida.
- Una volta completata la registrazione, scansiona l'immagine ora registrata per verificare la sovrapposizione delle ROI. Si tratta di ROI attivi in entrambe le sessioni di registrazione, che potrebbero essere utilizzati in ulteriori analisi.

Figura 6: Registrazione delle celle in modalità incrociata utilizzando il flusso di lavoro MIRA. (A) Immagine rappresentativa del flusso di lavoro di allineamento delle celle. L'immagine media dei dati a un fotone è mostrata a sinistra e quella dei dati a due fotoni è mostrata a destra. I punti di riferimento corrispondenti di entrambe le immagini vengono selezionati ed etichettati nel software da uno schema di colori casuale (cerchi rossi). (B) Le immagini allineate agli esempi che mostrano i due set di celle identificati, un fotone (viola) e due fotoni (verde), sono sovrapposte all'immagine media dei dati a due fotoni. (C) Immagine della regione contrassegnata con la casella bianca in (B), le celle allineate sono rappresentate qui come contorni verdi e viola sovrapposti. In tutti i pannelli, la barra della scala rappresenta 200 μm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Risultati
È stato mostrato il metodo di condurre l'imaging cronico del calcio multistrato in vivo della stessa popolazione neuronale per un periodo di diverse settimane, utilizzando modalità di imaging a uno o due fotoni, in condizioni di movimento libero e testa fissata. In questo caso, è stata dimostrata la capacità di identificare popolazioni neuronali corrispondenti nell'ambito dell'imaging a un fotone mentre l'animale esplorava un'arena aperta al buio (Figura 7A). Le tracce di calcio...
Discussione
Qui, abbiamo mostrato la capacità di osservare e confrontare direttamente i neuroni in condizioni di testa fissa e in movimento libero nelle stesse popolazioni neurali. Mentre abbiamo dimostrato l'applicazione nella corteccia visiva, questo protocollo può essere adattato a una moltitudine di altre aree cerebrali, sia aree corticali che nuclei profondi 24,25,26,27,28, così come altre acquisizioni di dati e configurazioni comportamentali
Divulgazioni
Gli autori dichiarano di non avere interessi finanziari concorrenti o conflitti di interesse.
Riconoscimenti
Ringraziamo la Sig.ra Charu Reddy e il Professor Matteo Carandini (Cortex Lab) per i loro consigli sul protocollo chirurgico e sulla condivisione del ceppo di topo transgenico. Ringraziamo il Dr. Norbert Hogrefe (Inscopix) per la sua guida e assistenza durante lo sviluppo dell'intervento. Ringraziamo la Sig.ra Andreea Aldea (Sun Lab) per la sua assistenza con la configurazione chirurgica e l'elaborazione dei dati. Questo lavoro è stato sostenuto dalla Moorfields Eye Charity.
Materiali
| Name | Company | Catalog Number | Comments |
| 0.9% Sodium Chloride solution for infusion (Vetivex 11) 250ml | Dechra | 20091607 | Saline for hydration and drug reconsitution |
| 18004-1 Trephine 1.8mm diameter bur | FST | 18004-18 | Drill bit |
| 1ml syringe | Terumo | MDSS01SE | 1ml syringe |
| 23G x 5/8 inch 6% LUER needle | Terumo | NN-2316R | 23G needle |
| 71000 Automated stereotaxic apparatus w/ built-in software | RWD | - | RWD |
| Absorbable Haemostatic Gelatin Sponge (10x10x10mm) | Surgispon | SSP-101010 | gel-foam |
| Alcohol pads 70% isopropyl alcohol | Braun | 9160612 | Alcohol pads |
| Aluminium foil | Any retailer | - | Foil to cover eyes during surgery |
| Articifical Cerebrospinal Fluid | Tocris Bioscience a Bio-Techne Brand | 3525/25ML | ACSF |
| Automated microinjection pump | WPI | 8091 | |
| Betadine solution (10% iodinated Povidone) 500ml | Videne/Ecolab | 3030440 | Betadine |
| Bruker Ultime 2Pplus (customised) | Bruker | - | Two-photon imaging system |
| Cardiff Aldasorber | Vet-Tech | AN006 | Anaesthesia absorber |
| CFI S Plan Fluor ELWD ADM 20XC | Nikon | MRH48230 | 20x objective lens |
| Compact Anaesthesia system - single gas - isoflurane K/F, with oxygen concentrator model: ZY-5AC and scavenging unit | Vet-Tech | AN001 | Compact anaesthesia system |
| Contec Prochlor | Aston Pharma | AP2111L1 | Disinfectant (hypochlorous acid) |
| Dexamethasone Sodium Phosphate Injection, USP, 4mg/ml, NDC: 0641-6145-25 | Hikma | Covetrus:70789 | Dexamethasone |
| Dissecting Knife, cutting edge 4mm, thickness 0.5mm, stainless steel | Fine Science Tools | 10055-12 | Knife for incisino of cortex |
| Dual-Sided, Non-Puncture Mouse & Neonatal Rat Ear Bars | Stoelting | 51649 | Ear bar |
| Dummy microscope | Inscopix | Dummy microscope | To help with implantation |
| Ethanol (100%) | VWR | 40-1712-25 | Used to make 70% ethanol |
| Fisherbrand Nitrile Indigo Disposable Gloves PPE Cat III | FischerScientific | 17182182 | Gloves |
| Homeothermic Monitor 50-7222-F | Harvard Apparatus | 50-7222-F | Homeothermic monitoring system/heating pad |
| Image processing software | ImageJ | - | Image processing software |
| Inscopix Data Processing Software (IDPS) | Inscopix | - | One-photon calcium imaging processing software |
| Insight Duals-232, S/N 2043 | InSight | Insight Spectra X3 | Two-photon imaging laser |
| IsoFlo 250ml 100% w/w inhalation | Zoetis | WM 42058/4195 | Isoflurane |
| Kwik-Sil Low Toxicity Silicone Adhesive | World Precision Intruments (WPI) | KWIK-SIL | Silicone adhesive |
| MICROMOT mains adapter NG 2/S, w/ Drill unit 60/E | PROXXON | NO 28 515 | Handheld drill |
| nVoke Integrated Imaging and Optogenetics System package | Inscopix | - | One-photon Imaging system and software |
| ProView Implant Kit | Inscopix | ProView Implant Kit | Dummy microscope, stereotaxic arm and attachment |
| ProView Prism Probe | Inscopix | 1050-002203 | Microprism lens |
| Rimadyl (50mg/ml) | Zoetis | VM 42058/4123 | Carprofen |
| Stereotaxis Microscope on Articulated arm with table clamp | WPI | PZMTIII-AAC | Microscope |
| Super-Bond Universal kit, SUN Medical | Prestige-Dental | K058E | Adhesive cement |
| Two-photon calcium image software | Suite2P | - | Two-photon calcium imaging processing software |
| Vapouriser | Vet-Tech | - | Isoflurane vapouriser |
| Xailin Lubricating Eye Ointment 5g | Xailin-Night | MLG/28/1551 | Ophthalmic ointment |
Riferimenti
- Denk, W., Strickler, J. H., Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science. 248 (4951), 73-76 (1990).
- Svoboda, K., Yasuda, R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 50 (6), 823-839 (2006).
- Dombeck, D. A., Khabbaz, A. N., Collman, F., Adelman, T. L., Tank, D. W. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 56 (1), 43-57 (2007).
- Vaziri, A., Emiliani, V. Reshaping the optical dimension in optogenetics. Curr Opin Neurobiol. 22 (1), 128-137 (2012).
- Holtmaat, A., et al. high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 4 (8), 1128-1144 (2009).
- Sun, Y. J., Sebastian Espinosa, J., Hoseini, M. S., Stryker, M. P. Experience-dependent structural plasticity at pre- and postsynaptic sites of layer 2/3 cells in developing visual cortex. Proc Natl Acad Sci U S A. 116 (43), 21812-21820 (2019).
- Andermann, M. L., Kerlin, A. M., Reid, R. C. Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci. 4, 3 (2010).
- Sofroniew, N. J., Flickinger, D., King, J., Svoboda, K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. Elife. 5, 14472 (2016).
- Puścian, A., Benisty, H., Higley, M. J. NMDAR-dependent emergence of behavioral representation in primary visual cortex. Cell Rep. 32 (4), 107970 (2020).
- Trachtenberg, J. T., et al. Long-term in vivo. imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 420 (6917), 788-794 (2002).
- Seaton, G., et al. Dual-component structural plasticity mediated by αCaMKII autophosphorylation on basal dendrites of cortical layer 2/3 neurones. J Neurosci. 40 (11), 2228-2245 (2020).
- Helmchen, F., Denk, W. Deep tissue two-photon microscopy. Nat Methods. 2 (12), 932-940 (2005).
- Andermann, M. L., et al. Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron. 80 (4), 900-913 (2013).
- Chia, T. H., Levene, M. J. Microprisms for in vivo multilayer cortical imaging. J Neurophysiol. 102 (2), 1310-1314 (2009).
- Low, R. J., Gu, Y., Tank, D. W. Cellular resolution optical access to brain regions in fissures: Imaging medial prefrontal cortex and grid cells in entorhinal cortex. Proc Natl Acad Sci U S A. 111 (52), 18739-18744 (2014).
- Buxhoeveden, D. P., Casanova, M. F. The minicolumn hypothesis in neuroscience. Brain. 125, 935-951 (2002).
- Chen, S., et al. Miniature fluorescence microscopy for imaging brain activity in freely-behaving animals. Neurosci Bull. 36 (10), 1182-1190 (2020).
- Gulati, S., Cao, V. Y., Otte, S. Multi-layer cortical Ca2+ imaging in freely moving mice with prism probes and miniaturized fluorescence microscopy. J Vis Exp. (124), e55579 (2017).
- Resendez, S. L., et al. Visualization of cortical, subcortical, and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat Protoc. 11 (3), 566-597 (2016).
- Guo, Z. V., et al. Procedures for behavioral experiments in head-fixed mice. PLoS One. 9 (2), 88678 (2014).
- Wekselblatt, J. B., Flister, E. D., Piscopo, D. M., Niell, C. M. Large-scale imaging of cortical dynamics during sensory perception and behavior. J Neurophysiol. 115 (6), 2852-2866 (2016).
- Pnevmatikakis, E. A., et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron. 89 (2), 285-299 (2016).
- Zhou, P., et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife. 7, 28728 (2018).
- Beckmann, L., et al. Longitudinal deep-brain imaging in mouse using visible-light optical coherence tomography through chronic microprism cranial window. Biomed Opt Express. 10 (10), 5235-5250 (2019).
- Wenzel, M., Hamm, J. P., Peterka, D. S., Yuste, R. Reliable and elastic propagation of cortical seizures in. Cell Rep. 19 (13), 2681-2693 (2017).
- Heys, J. G., Rangarajan, K. V., Dombeck, D. A. The functional micro-organization of grid cells revealed by cellular-resolution imaging. Neuron. 84 (5), 1079-1090 (2014).
- Barson, D., Hamodi, A. S. Simultaneous mesoscopic and two-photon imaging of neuronal activity in cortical circuits. Nat Methods. 17 (1), 107-113 (2020).
- Paquelet, G. E., et al. Single-cell activity and network properties of dorsal raphe nucleus serotonin neurons during emotionally salient behaviors. Neuron. 110 (16), 2664-2679 (2022).
- Yang, Q., et al. Transparent microelectrode arrays integrated with microprisms for electrophysiology and simultaneous two-photon imaging across cortical layers. bioRxiv. , (2022).
- Priestley, J. B., Bowler, J. C., Rolotti, S. V., Fusi, S., Losonczy, A. Signatures of rapid plasticity in hippocampal CA1 representations during novel experiences. Neuron. 110 (12), 1978-1992 (2022).
- Zong, W., et al. Miniature two-photon microscopy for enlarged field-of-view, multi-plane and long-term brain imaging. Nat Methods. 18 (1), 46-49 (2021).
- Engelbrecht, C. J., et al. Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express. 16 (8), 5556-5564 (2008).
- Suzuki, M., Aru, J., Larkum, M. E. Double-μ Periscope, a tool for multilayer optical recordings, optogenetic stimulations or both. Elife. 10, 72894 (2021).
- Stibůrek, M., et al. 110 μm thin endo-microscope for deep-brain in vivo observations of neuronal connectivity, activity and blood flow dynamics. Nat Commun. 14 (1), 1897 (2023).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati