
Method Article
Monitoring On-Target Signaling Responses in Larval Zebrafish - Z-REX Unmasks Precise Mechanisms of Electrophilic Drugs and Metabolites
W tym Artykule
Podsumowanie
Zebrafish targeting reactive electrophiles and oxidants (Z-REX) is a chemical biology-based method for the investigation of reactive small-molecule signaling. This technique can be applied to live fish of different developmental stages. Here, we couple standard assays in zebrafish with Z-REX for signaling pathway analysis.
Streszczenie
Reactive metabolites and related electrophilic drugs are among the most challenging small molecules to study. Conventional approaches to deconstruct the mode of action (MOA) of such molecules leverage bulk treatment of experimental specimens with an excess of a specific reactive species. In this approach, the high reactivity of electrophiles renders non-discriminate labeling of the proteome in a time- and context-dependent manner; redox-sensitive proteins and processes can also be indirectly and often irreversibly affected. Against such a backdrop of innumerable potential targets and indirect secondary effects, linking phenotype to specific target engagement remains a complex task. Zebrafish targeting reactive electrophiles and oxidants (Z-REX)-an on-demand reactive-electrophile delivery platform adapted for use in larval zebrafish-is designed to deliver electrophiles to a specific protein of interest (POI) in otherwise unperturbed live fish embryos. Key features of this technique include a low level of invasiveness, along with dosage-, chemotype-, and spatiotemporally-controlled precision electrophile delivery. Thus, in conjunction with a unique suite of controls, this technique sidesteps off-target effects and systemic toxicity, otherwise observed following uncontrolled bulk exposure of animals to reactive electrophiles and pleiotropic electrophilic drugs. Leveraging Z-REX, researchers can establish a foothold in the understanding of how individual stress responses and signaling outputs are altered as a result of specific reactive ligand engagement with a specific POI, under near-physiologic conditions in intact living animals.
Wprowadzenie
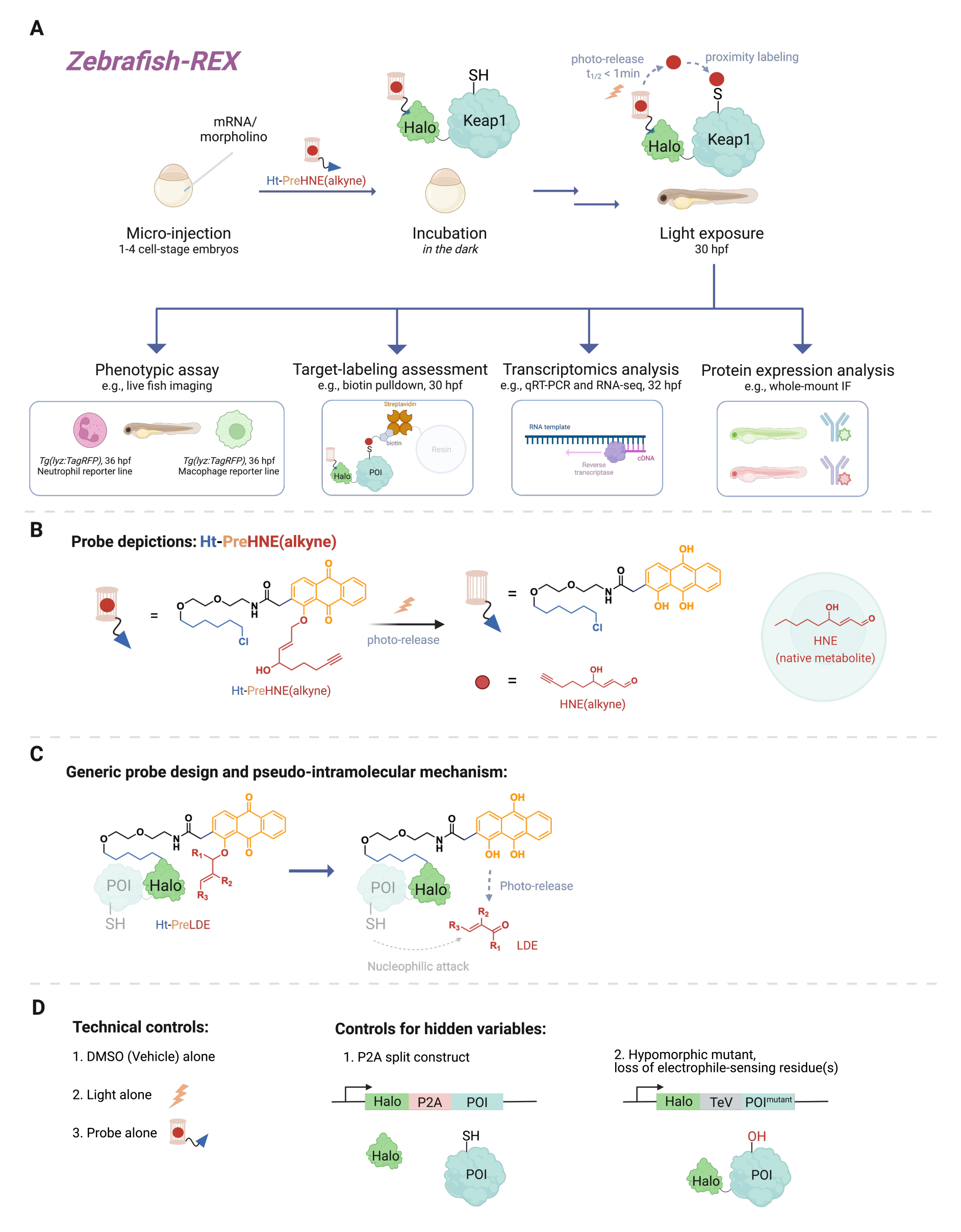
A myriad of cellular signaling events involve reactions between small reactive molecules (endogenously produced in the cell or xenobiotics/xenometabolites, such as drugs) and their protein target. In many instances, a substoichiometric level of such covalent binding events can trigger cellular responses, leading to changes in, for instance, development, metabolism, apoptosis, and/or immune response1. However, deconstructing the mode of action (MOA) by linking specific binding events to their phenotypic consequences has proven challenging. Traditional bolus dosing methods that involve the introduction of high concentrations of the reactive species often result in a multitude of proteins being modified, as well as excessive toxicity to the model organism2. Such conditions are far from ideal. A method was developed to resolve these issues in cell culture using precision localized electrophile delivery in a native cellular context, named T-REX (targetable reactive electrophiles and oxidants)3. In the intervening years, the focus has turned to experiments in whole organisms, which allow the opportunity to study proteins in specific cellular contexts in non-transformed cells. Thus, we have extended the technique to be compatible with several models, including Danio rerio embryo models. Herein, we present Z-REX (zebrafish targeting reactive electrophiles and oxidants) (Figure 1).
To understand Z-REX, this article first presents REX technologies and their underlying concepts. At their core, these techniques model endogenous physiological reactive electrophilic species (RES) signaling by mimicking how natural electrophiles are locally produced in vivo with spatiotemporal precision. The protein of interest (POI) is expressed as a fusion construct to Halo; the latter anchors the tissue-permeable and inert probe bearing the photocaged RES in a 1:1 stoichiometry. One such endogenous RES is 4-hydroxynonenal (HNE hereafter), which is photocaged in the probe Ht-PreHNE. In many instances, we use an alkyne-functionalized version of HNE [i.e., HNE(alkyne)], which has essentially identical biological properties to HNE, but can be labeled by click chemistry. The probe, which is also functionalized with a chloroalkane for reactivity with Halo, is referred to as Ht-PreHNE(alkyne). The complex of the Halo-POI fusion and the probe thus formed allows proximal delivery of RES to the fused POI upon irradiation with UV light. If the POI reacts rapidly with the liberated RES, the resulting covalent labeling of the POI with RES allows us to identify kinetically-privileged cysteines.
Z-REX takes the aforementioned advantages of REX technologies and applies them broadly to study specific signaling pathways in live fish. This protocol has been optimized for zebrafish (D. rerio), since they are genetically-tractable vertebrate organisms that are transparent during development, and thus ideal for opto-chemical/-genetic techniques like REX technologies. Nevertheless, a similar strategy is also likely to work well on other genetically-tractable fish species, since the broad applicability of the method is due to the pseudo-intramolecular mechanism of lipid-derived electrophile (LDE) delivery. Indeed, the procedure is highly biocompatible, since fish can be treated with the Z-REX photocaged-electrophile [e.g., Ht-PreHNE(alkyne)] for at least 48 h without any noticeable impacts on development. A similar protocol functions in C. elegans4,5.
The protocol first describes the use of mRNA injection to produce a transient expression of a non-native Halo-POI fusion construct in embryonic zebrafish models, 1-1.5 days post-fertilization (dpf). This results in the expression of the ectopic protein in the majority of cells within the fish (hereafter referred to as 'ubiquitous'), rather than in specific tissues or locales; however, the data shows that cell-specific effects can be observed in certain cases. Following injection, the embryos are incubated with a low concentration [0.3-5 µM Ht-PreHNE(alkyne)] of the probe for up to 30.5 h post fertilization (hpf). Then, at a user-prescribed time, delivery of the RES to the POI within fish is achieved by photouncaging for 2-5 min. Following photouncaging of the RES, a variety of downstream phenotypic assays can be performed over the next 2-10 h: 1) live imaging of reporter lines (Figure 2A); 2) target-labeling assessment by western blot analysis (Figure 3); 3) transcriptomic analysis (Figure 4); or 4) whole-mount immunofluorescence (Figure 5).
As an example of live imaging of reporter lines, Z-REX is demonstrated in tandem with live imaging of fish lines, Tg(lyz:TagRFP) and Tg(mpeg1:eGFP), to measure how RES modification of a specific electrophile-sensor POI (namely Keap1) decreases neutrophil and macrophage levels, respectively, with no observable effects on other cells in the fish. However, we have shown previously that the POI-labeling and the consequential pathway signaling from T-REX studies can be reproduced using Z-REX for several proteins: Akt36, Keap17, and Ube2v26. Overall, with Z-REX, scientists can study the consequence(s) of covalent modification of POIs by RES in the context of several complex redox pathways. This technique is primed to pinpoint targets and their functional residues for covalent drug design and novel drug mechanisms in a more contextually-relevant whole animal model.
Protokół
Zebrafish husbandry and handling procedures at Cornell University (United States) were performed following the guidelines of the National Institutes of Health (NIH) and approved by the Institutional Animal Care and Use Committee (IACUC). Zebrafish husbandry and handling procedures at the zebrafish unit of the Swiss Federal Institute of Technology Lausanne (EPFL, Switzerland) were performed following Animal Welfare Act SR 455 and Animal Welfare Ordinance SR 455.1, with cantonal veterinary authorization VD-H23.
NOTE: In this protocol, Tg(lyz:TagRFP) and Tg(mpeg1:EGFP) fish lines expressing Halo-TeV-Keap1 are used to demonstrate Z-REX. The method can be extended to other proteins of interest, transgenic reporter fish lines, and downstream biological assays. Refer to Supplementary Table 1 for the buffers used in this study. All the reagents, instruments, equipment, antibodies, plasmids, zebrafish strains and equipment are listed in the Table of Materials.
1. mRNA preparation
- Prepare Halo-TeV-Keap1-2xHA and Halo-2xHA-P2A-Keap1-2xHA mRNA using the mMessage mMachine SP6 in vitro transcription kit.
NOTE: Follow the manufacturer's instructions and perform 40 µL scale reactions for each mRNA. Redissolve the mRNA pellet in 10 µL of nuclease-free water. - Assess the mRNA quality and measure the concentration by microvolume spectrophotometer and agarose gel electrophoresis. An mRNA of good quality should have an A260/A280 ratio of around or above 2.0.
- Dilute the mRNA to 1-1.5 mg/mL with nuclease-free water.
- Aliquot the mRNA solution (1-2 µL per tube), and store the aliquots at -80 °C.
2. Producing fish embryos
- Option 1: Producing wild-type (WT) fish embryos.
- Set up 10 fish crossing pairs in 10 separate tanks, with each tank containing a divider between the male and female parent fish.
NOTE: A total of 10 crossing pairs typically provides a sufficient number of embryos for the assays. The number of crossing pairs can be adjusted according to the experiment design/need and the fecundity of the parent fish. - The following morning, after setting up the injector, remove the dividers in five of the tanks. Wait for 30 min for the fish to mate.
- Move the parent fish to another tank, collect embryos by passing the tank water through a strainer, and then rinse out the embryos from the strainer into a 10 cm Petri dish. These embryos will be used for the first round of injections.
NOTE: If the egg quality from a certain batch is poor (e.g., eggs are opaque due to aggregation of proteins), do not pool them with the other embryos. - (Optional) Perform similar procedures as described in steps 2.1.2-2.1.3 on the other five tanks for the next round of injection.
NOTE: It is best only to perform one round of injection, to minimize the age difference between embryos. However, should more embryos than can be injected in one lot be required, performing two rounds of injection is recommended to ensure the embryos remain at a 1-4-cell stage during mRNA injection. The number of injection rounds can be adjusted according to the operator's injection skill and experiment design. However, it is suggested to perform the whole procedure (from the removal of the first divider to the injection of the last embryo) within 2 h. A large age difference across embryos may impair the reliability and reproducibility of the experiment results.
- Set up 10 fish crossing pairs in 10 separate tanks, with each tank containing a divider between the male and female parent fish.
- Option 2: Producing heterozygous transgenic neutrophil/macrophage reporter fish embryos.
- Set up 10 fish crossing pairs in 10 separate tanks, with each tank inserted with a divider between male and female parent fish: WT fish versus Tg(lyz:TagRFP), or WT fish versus Tg(mpeg1:eGFP).
NOTE: Avoid in-crosses between heterozygous transgenic fish, which may affect downstream fluorescent readouts as homozygous reporter fish show a higher fluorescent signal compared to heterozygous fish. Transgenic reporter lines and WT embryos can be easily distinguished when imaging. Having a mixture of WT and heterozygous embryos in the same pool is not a problem. lyz:TagRFP reports neutrophils, and mpeg:eGFP reports macrophages. This protocol can be applied to other reporter fish lines as well. - Follow steps 2.1.2-2.1.4.
- Set up 10 fish crossing pairs in 10 separate tanks, with each tank inserted with a divider between male and female parent fish: WT fish versus Tg(lyz:TagRFP), or WT fish versus Tg(mpeg1:eGFP).
3. Microinjector setup
- Turn on the air source, set the back pressure to 0.2-0.5 psi, and set the injection pressure to 25-30 psi. The specific range of pressure shown is typically recommended.
NOTE: It is essential to have a stable back pressure to prevent fish media backflow into the needle. When calibrating the injection volume in step 3.8, only the injection time should be changed. Do not change the injection pressure in the following steps; low injection pressure can lead to injection failure due to surface and interfacial tension, whereas high injection pressure can damage embryos. - Clean the equipment and injection platform with RNase decontamination solution.
NOTE: RNase, which degrades mRNA, could come from the operator or the equipment. It is necessary to perform the cleanup before the experiment, and wear gloves. - (Optional) If co-injecting mRNA and morpholino, premix the two in a 0.2 mL tube.
NOTE: Z-REX works well by using Halo-TeV-Keap1-2xHA mRNA solution with a concentration of 250-1500 ng/µL. Several morpholinos have also been used in zebrafish, and the optimal concentrations have been reported7. If using a morpholino with an unpublished sequence, the operator should first assess the toxicity and gene knockdown efficiency of the morpholino before using it in Z-REX. - Transfer 1-2 µL of mRNA (and/or morpholino where applicable7) into an injection needle with a micro-loader pipette tip.
NOTE: If preparing needles with a Flaming/Brown micropipette puller, the setup is as follows. Heat: 520 units; pull strength: 60 units; velocity: 70 units; delay: 155 units; pressure: 550 units; ramp: 530 units. - Install the needle on the microinjection manipulator.
NOTE: Back pressure from the air source should push the mRNA(/morpholino) solution to the needle tip. - Use sharp forceps or a razor blade to break the needle tip, creating a suitable opening for injection.
- Submerge the needle tip in mineral oil on a stage micrometer.
- Apply two or three injection pulses to remove air bubbles in the tip.
- Calibrate the drop size to 2 nL by changing the injection time.
NOTE: This is best performed by injecting into mineral oil (which mimics the viscosity of the yolk sac) laid upon a hemocytometer. Using a microscope, use the gridlines of the hemocytometer to estimate the size of the droplet formed during injection, and adjust the injection time accordingly. Although phenol red dye is sometimes used, the need for it in the mRNA injection procedure described here has not been observed.
4. Microinjection
- Fill an injection plate with fresh 10% Hank's balanced salt solution (HBSS) medium, and align the embryos in the grooves of the plate with blunt forceps.
NOTE: The injection plate is prepared with 2% agarose in 10% HBSS medium; the grooves are formed using a plastic mold. - Submerge the needle tip in 10% HBSS medium in the injection plate.
- Apply two or three injection pulses to remove air bubbles in the tip.
- For each injection, penetrate the chorion and yolk sac in a single move and apply an injection pulse. This injected liquid can be seen right after the injection as a small spheroid within the yolk sac. This small spheroid dissipates relatively rapidly. Repeat this step for other embryos, until a sufficient number of injected embryos has been obtained.
NOTE: The survival rate of embryos (both injected and non-injected) typically varies from 50%-90%. Aim to inject double the number of embryos needed for each control/experimental group. In the biotin pull-down assay, 100-140 viable embryos are required for each condition. In the qRT-PCR assay, five viable embryos are recommended for each condition. The sample size for live fish imaging and whole-mount immunofluorescence staining assay is user-defined; having at least 20 viable embryos per condition is recommended to yield good statistical power in the analysis. - Rinse out the injected embryos to a new 10 cm Petri dish containing fresh 10% HBSS media.
NOTE: The embryos can be easily rinsed out from the grooves using a squirt bottle. - Pool the non-injected embryos into another plate.
NOTE: The non-injected embryos can serve as quality controls for the health of the fish, baseline protein expression, background fluorescence levels, etc., as needed. If the injection procedure worked well, and injected mRNA/morpholino is not lethal to the embryos, injected and non-injected embryos should have similar viability.
5. Z-REX
- Distribute the injected embryos into 10 cm dishes, according to the experiment setup (i.e., numbers of control/experiment groups).
- In a dark room with red-light illumination, replace the media with 30 mL of 10% HBSS with or without 1 µM Ht-PreHNE(alkyne).
NOTE: Ht-PreHNE(alkyne) is a light-labile compound. Embryos should be kept in the dark in the following steps. - Incubate the embryos at 28.5 °C in the dark.
- At 30.5 hpf, in a dark room, replace the media with a fresh 30 mL of 10% HBSS.
NOTE: When replacing the medium, remove as much of the old medium as possible. This is critical for removing the unbound/excess amount of Ht-PreHNE(alkyne) from the embryos. - Incubate the embryos at 28.5 °C in the dark for 30 min.
- Repeat steps 5.4-5.5 two more times.
- Turn on the UV lamp (365 nm, 3 mW/cm2) for 5 min to prewarm the lamp.
NOTE: The lamp prewarming step must be performed before step 5.8. The lamp power is lower/unstable in the first few minutes after being turned on. The lamp power should be measured by a UV meter regularly. - At 32 hpf, expose the embryos to UV light.
- Option 1: For downstream readouts, such as live fish imaging, whole-mount immunofluorescence staining, in-gel fluorescence analysis (click coupling with Cy5 azide), RNA-seq, or qRT-PCR, expose the embryos to UV light for 3 min, swirling the plates every 30 s.
- Option 2: For downstream readouts, such as biotin pull-down assay, expose the embryos to UV light for a maximum of 5 min (and minimum of 3 min), swirling the plates every 30 s, and chill the plates on ice for 1 min.
NOTE: If using different probes, the light exposure time needs to be optimized, depending on the t1/2 of photouncaging for a given photocaged electrophile probe and light source deployed. t1/2photouncaging can be determined using known procedures8. For Ht-PreHNE(alkyne), t1/2 is < 1 min3; thus, the indicated time above is sufficient.
6. Downstream assays
- Option 1: Phenotypic assay. Live imaging of transgenic neutrophil/macrophage reporter
fish lines, Tg(lyz:TagRFP) and Tg(mpeg1:eGFP) (Figure 2)- Anesthetize the embryos by incubation at 4 °C for 10 min.
NOTE: It is recommended to have at least 20 viable embryos per condition. - Remove the unfertilized/dead embryos from the plate.
NOTE: The unfertilized/dead embryos are cloudy/non-transparent, and can be visually identified. If a high death rate is seen, check back to the injection procedure or try to reduce the concentration of mRNA or morpholino. - Dechorionate the embryos with sharp forceps. Hold the chorion with one pair of forceps, without touching the larval fish, and use the other pair of forceps to rip off the chorion.The embryo is fragile. Only touch the chorion when performing dechorionation.
NOTE: It is common, especially in beginners, to damage some embryos during dechorionation. Therefore, always have more embryos than the minimum needed. - Mount the embryos on a 2% agarose plate (prepared with 10% HBSS medium) and image the embryos with a stereomicroscope (bright field and respective fluorescent channels) (Figure 2A, C, G).
NOTE: After Z-REX [combination of Halo-TeV-Keap1-2xHA mRNA injection and Ht-PreHNE(alkyne) treatment], the depletion of neutrophils (lyz:TagRFP) was found to be most significant at 36 hpf (4 h post Z-REX), whereas the reduction of macrophages (mpeg1:eGFP) was most significant at 34 hpf (2 h post Z-REX) (Figure 2E,F). Other time points can be used if using different reporter lines, mRNA/morpholino, or probes. The exposure time and/or gain must be optimized for visualizing single cells or the specific (ultra)structures desired. - Count the neutrophil/macrophage number of each fish by ImageJ (NIH) (Figure 2B, D-F, H). Use the Freehand Selection tool in ImageJ to circle the whole fish, and use the option Find Maxima to count the fluorescent cells.
- Anesthetize the embryos by incubation at 4 °C for 10 min.
- Option 2: Target-labeling assessment. Biotin azide-click coupling and biotin pull-down assay (Figure 3)
- Anesthetize the embryos by incubation at 4 °C. This usually takes 10 min.
NOTE: To obtain enough fish lysate, 100-140 viable embryos are required for each condition. - Remove the unfertilized/dead embryos from the plate.
- Perform dechorionation and deyolking. Perform the two manipulations by holding the chorion with a pair of sharp forceps, using the other pair of forceps to penetrate the yolk sac, and then separating the yolk sac while removing the chorion to allow the yolk proteins to come out.
NOTE: It is critical to remove the yolk proteins in this step. The abundant yolk proteins in the sample interfere with later analysis. - Transfer the deyolked embryos into a 1.5 mL tube.
NOTE: Swirl the plate to center the deyolked embryos, which facilitates collection. The lighter chorion debris winnows away during this process. - After the embryos settle down to the tube bottom, remove the supernatant, and add 1 mL of chilled HEPES-buffered saline (pH 7.6).
- Repeat step 6.2.5 two more times.
- (Optional) If not intending to proceed with the next step immediately, remove the buffer, flash freeze the samples in liquid nitrogen, and store them at -80 °C.
NOTE: Deyolked fish pellets can be flash-frozen in liquid nitrogen and stored at -80 °C. Flash-frozen samples can be stored at -80 °C for up to 2 weeks. - Resuspend the fish pellets in lysis buffer.
NOTE: Lysis buffer (pH 7.6) is composed of 50 mM HEPES, 100 mM NaCl, 1% Triton X-100, 0.3 mM TCEP, 2x Roche cOmplete Mini EDTA-free protease inhibitors, and 0.1 mg/mL trypsin inhibitor from soybean. One embryo gives around 2 µg of lysate. Use 100 µL of lysis buffer for every 120 embryos. Two Roche cOmplete Mini EDTA-free protease inhibitors and trypsin inhibitors from soybean should be added to the lysis buffer just before use. - Add 20% v/v zirconia beads to the tube.
- Vortex for 20 s, flash freeze in liquid nitrogen, and thaw in a 37 °C water bath.
- Repeat step 6.2.10 another two times.
- Centrifuge the solution at 21,000 x g at 4 °C for 10 min.
- Transfer the supernatant to a new, prechilled 1.5 mL tube.
- Measure the protein concentration by Bradford assay.
- Dilute the lysate to 1 mg/mL.
- For each condition, transfer 170 µg of lysate into a 2 mL tube.
- Mix the lysate from step 6.2.16 with 0.2 mg/mL TeV protease(S219V), and incubate the solution at 37 °C for 30 min.
NOTE: For non-TeV protease-treated groups, simply mix the lysate with an equal voloume of lysis buffer to the TeV protease solution used in other groups. - Prepare a 10x master mix for the biotin-azide click reaction: 10% w/v SDS, 10 mM CuSO4, 1 mM Cu(TBTA), 1 mM biotin-azide, and 20 mM TCEP.
NOTE: Add TCEP to the mix just before step 6.2.19. - Add 8.5 µL of t-BuOH and 17 µL of 10x click reaction master mix to the (TeV protease-digested) lysate from step 6.2.17. Vortex, centrifuge, and incubate the solution at 37 °C for 15 min.
- Add another 1 mM TCEP to the solution, and then vortex, centrifuge, and incubate the solution at 37 °C for another 15 min. The incubation time for steps 6.2.19-6.2.20 is 30 min in total.
NOTE: This supplement of TCEP, a reducing reagent for generating Cu(I), improves the click reaction efficiency. - Add 600 µL of -20 °C ethanol to each tube, vortex the solution, and incubate it at -80 °C overnight.
NOTE: The samples can be stored at -80 °C for 1 week; if not, proceed with the next step immediately. - Centrifuge the solution at 21,000 x g at 4 °C for 1 h.
NOTE: A pellet should form in the bottom of the tube after centrifuging, which is the desired fraction. - Remove the supernatant, add 1 mL of -20 °C ethanol, vortex, and centrifuge the solution at 21,000 x g at 4 °C for 10 min.
- Repeat step 6.2.23.
- Remove the supernatant, add 1 mL of -20 °C acetone, vortex, and centrifuge the solution at 21,000 x g at 4 °C for 10 min.
- Remove the supernatant. Allow excess residual acetone to evaporate, though not completely to dryness.
- Resuspend the pellet in 100 µL of resuspension buffer (8% w/v lithium dodecyl sulfate [LDS], 1 mM EDTA in 50 mM HEPES saline, pH 7.6), vortex for 15 s, and sonicate until the pellet is dissolved.
- Centrifuge the solution at 21,000 x g at room temperature (RT) for 5 min.
- Transfer the supernatant to a new 2 mL tube, and add 1.5 mL of 50 mM HEPES saline, pH 7.6.
NOTE: The final concentration of LDS in this step is 0.5%. Higher LDS concentrations may undermine the pull-down efficiency. Thus, although increasing the LDS concentration can aid the reduction of non-specific binding, it may also reduce the efficiency of the pulldown. Accordingly, it is recommended not to change the LDS concentration at this step. - Collect the "input" sample (Figure 3): transfer 30 µL of 1 mg/mL lysate to a new 1.5 mL tube, and add 10 µL of 4x Laemmli sample buffer containing 6% β-Mercaptoethanol (BME). Flash freeze the solution, and store it at -80 °C.
- Transfer 100 µL of bed-volume streptavidin high-capacity resin to a new 2 mL tube. Add 1 mL of 0.5% LDS in 50 mM HEPES saline (pH 7.6), centrifuge at 1,500 x g at RT for 2 min, and remove the supernatant. Repeat the wash with another 1 mL of 0.5% LDS in 50 mM HEPES saline (pH 7.6).
- Transfer the solution from step 6.2.29 to the tube containing prewashed streptavidin high-capacity resin from step 6.2.31, and incubate the solution on an end-over-end mixer at RT for 4-6 h.
- Centrifuge the mixture at 1,500 x g at RT for 2 min, take 30 µL of the supernatant, and mix it with 10 µL of 4x Laemmli sample buffer containing 6% BME for the "flowthrough" sample. Then, remove the remaining supernatant.
NOTE: "Flowthrough" samples can be analyzed by western blotting to check the streptavidin pull-down efficiency. It is essential to remove as much of the supernatant as possible to wash out the unbound proteins. First, remove most of the supernatant using a P-1000 pipette, and then remove the remaining supernatant using a P-20 pipette with a gel loading tip. - Add 1 mL of 0.5% LDS in 50 mM HEPES saline (pH 7.6) to the resin, and incubate the mixture for 30 min at RT with end-over-end rotation.
- Centrifuge the mixture at 1,500 x g at RT for 2 min, and remove the supernatant.
NOTE: Typically, 0.5% LDS is enough for removing most non-specific binding proteins. If non-specific binding signals are still seen in the later analysis, the LDS concentration in the wash buffer can be augmented. - Repeat step 6.2.34-6.2.35 two more times.
- Add 40 µL of 2x Laemmli sample buffer containing 6% BME to the resin.
- Elute the bound proteins by incubating the mixture at 98 °C for 5 min.
- Centrifuge the mixture at 21,000 x g at RT for 5 min, and transfer the supernatant to a new 1.5 mL tube. This is the "elute" sample.
NOTE: Directly loading solution containing resin from step 6.2.38 into the gel may affect SDS-PAGE analysis. - Load 20 µL into each well of 10-lane 10% polyacrylamide gel, and run gel electrophoresis.
NOTE: Run the gel at a lower voltage (120 V) until the dye front reaches the resolving gel, and change the voltage to 170 V. Stop the program after the dye front comes out. - Perform western blotting with anti-HA, anti-Halo, or other antibodies detecting housekeeping proteins (Figure 3).
- Anesthetize the embryos by incubation at 4 °C. This usually takes 10 min.
- Option 3: Transcriptomic analysis. RNA-seq and qRT-PCR (Figure 4)
NOTE: It is strongly recommended to use embryos laid within 15 min of one another for this assay. The age difference of the embryos significantly affects the assay results.- Anesthetize the embryos by incubating them at 4 °C for 10 min, 2 h after Z-REX.
- Perform dechorionation with sharp forceps (step 6.1.3).
- (Optional) Perform segmentation with forceps (e.g., separate the head from the tail) if different segments are to be analyzed separately.
- If extracting RNA from a whole embryo, transfer three to five embryos into a 1.5 mL tube. If extracting RNA from the head or tail, transfer 10-12 dissected segments into a 1.5 mL tube.
NOTE: It is recommended to perform the experiment with three to five biological replicates. - Add 1 mL of TRIzol reagent and glass beads to the tube.
NOTE: It was found that glass beads work better than zirconia beads for RNA extraction. - Vortex the mixture for 30 s.
NOTE: If not proceeding with the next step immediately, the solution can be stored at -80 °C for 1−3 weeks. - Extract the RNA according to the manufacturer's instructions.
- Assess the RNA quality and concentration by microvolume spectrophotometer and agarose gel electrophoresis.
NOTE: RNA of good quality should have an A260/A280 ratio of around or above 2.0. - Either submit the RNA for sequencing, or treat 1 µg of RNA with amplification-grade DNase I, and reverse transcribe using superscript III reverse transcriptase and oligo-(dT)20. Perform this step according to the manufacturer's instructions.
- Perform qRT-PCR and analyze the data by the ΔΔCT method9 (Figure 4B-D).
- Option 4: POI expression and colocalization analysis. Whole-mount immunofluorescence staining assay (Figure 5)
NOTE: Formaldehyde-fixed embryos are fragile. Avoid vigorous shaking, and handle with care.- Dechorionate the embryos by following steps 6.1.1-6.1.3.
- Transfer the embryos to a 1.5 mL tube.
NOTE: Each tube should have an equal number of embryos, and no more than 40 embryos. - After the embryos settle to the bottom of the tube, remove the supernatant, and add 1 mL of phosphate buffered saline (PBS) (pH 7.6).
- Repeat step 6.4.3 one more time.
- Remove the supernatant, add 1 mL of 4% formaldehyde in PBS (pH 7.6), and incubate the tube at 4 °C overnight with gentle rocking.
NOTE: The samples in formaldehyde solution can be stored at 4 °C for 1 week. - Remove the supernatant, add 1 mL of -20 °C methanol, and incubate the tube on its side at -20 °C for at least 18 h.
NOTE: The samples can be stored at -20 °C for 1 month or more. - Remove the supernatant, and add 1 mL of PDT buffer (0.3% v/v Triton X-100, 0.1% v/v Tween-20, and 1% v/v dimethyl sulfoxide [DMSO] in PBS buffer).
- Repeat step 6.4.7, and incubate the tube at RT for 30 min with gentle rocking.
- Remove the supernatant, add 1 mL of blocking buffer (10% v/v heat-inactivated fetal bovine serum [FBS], 2% w/v bovine serum albumin [BSA], and 0.1% v/v Tween-20 in PBS buffer), and incubate the tube at RT for 1 h with gentle rocking.
- Remove the supernatant, and add 200 µL of primary antibody solution (diluted in blocking buffer).
- Remove the supernatant, add 500 µL of primary antibody solution (diluted in blocking buffer), and incubate the tube at 4 °C overnight with gentle rocking.
NOTE: If using a new primary antibody, it is worth including some samples without primary antibody staining to serve as negative controls. However, ideally, morpholino-knockdown or engineered gene-knockout embryos, or embryos in which expression of the target protein has been stimulated, are more reliable means to validate the antibody. - Remove the supernatant, add 1 mL of PDT buffer, and incubate the tube at RT for 30 min with gentle rocking.
- Repeat step 6.4.12.
- Remove the supernatant, add 1 mL of blocking buffer, and incubate the tube at RT for 1 h with gentle rocking.
NOTE: The samples should be protected from light after this step, to prevent photobleaching of the fluorophore conjugated on the secondary antibody. - Remove the supernatant, and add 200 µL of secondary antibody solution (diluted in blocking buffer).
- Remove the supernatant, add 500 µL of secondary antibody solution (diluted in blocking buffer), and incubate the tube at RT for 1.5 h with gentle rocking.
- Remove the supernatant, add 1 mL of PDT buffer, and incubate the tube at RT for 30 min with gentle rocking.
- Repeat step 6.4.17.
- Mount the embryos on a 2% agarose plate (made with PBS, pH 7.6) and image the embryos with a stereomicroscope (bright field and respective fluorescent channels) (Figure 5A,B,D,F).
NOTE: If using the Leica M165 FC fluorescence stereomicroscope, use a magnification of 25x to obtain images with good resolution. - Quantify/analyze the fluorescent signal intensity by ImageJ (NIH). Use the Freehand Selection tool in ImageJ to quantify the signal in the region of interest.
Wyniki
Live imaging of Z-REX-treated transgenic neutrophil/macrophage reporter fish, Tg(lyz:TagRFP) and Tg(mpeg1:EGFP). Induction of neutrophil/macrophage apoptosis through Keap1 HNEylation. (See also Figure 2). The effect of electrophile labeling of Keap1 on neutrophil and macrophage levels was assessed by injecting heterozygous transgenic embryos derived from Tg(lyz:TagRFP) or Tg(mpeg1:EGFP) with mRNA encoding Halo-Keap1, and then treating with Ht-PreHNE(alkyne). Following the procedures for step 6.1-downstream assay Option 1-HNE(alkyne) was liberated and Keap1 was labeled. Neutrophil and macrophage levels were assessed by live imaging of reporter lines, Tg(lyz:TagRFP) and Tg(mpeg1:eGFP), respectively. The level of both cell types decreased by 30%-40% after Z-REX treatment, in which HNE was delivered to Keap1. On the contrary, no loss of neutrophils or macrophages was seen in Z-REX technical control groups [without light and Ht-PreHNE(alkyne), light alone, or Ht-PreHNE(alkyne) alone] (Figure 1D and Figure 2A-D).
The induction of neutrophil/macrophage apoptosis indicated successful HNE delivery to Keap1 through Z-REX. Details for the pathway analysis and apoptosis mechanism have been published5. To account for off-target effects of HNE(alkyne), several controls were used. (1) Under the same experimental conditions, instead of Halo-TeV-Keap1 mRNA, embryos were injected with Halo-P2A-Keap1 mRNA. P2A linker allowed the Halo and Keap1 proteins to be expressed independently. In this scenario, HNE(alkyne) released from Halo could not label Keap1, as it was no longer proximal to Halo (Figure 1D); hence, the apoptosis signaling pathway was not triggered. No changes in macrophage or neutrophil levels were observed in this group (Figure 2A,B). (2) The same experimental conditions were performed using mRNA encoding Halo-TeV-Keap1(C151S,C273W,C288E), a mutant of Keap1 that does not respond to HNE(alkyne) (Figure 1D). No changes in macrophage or neutrophil levels were observed (Figure 2G,H).
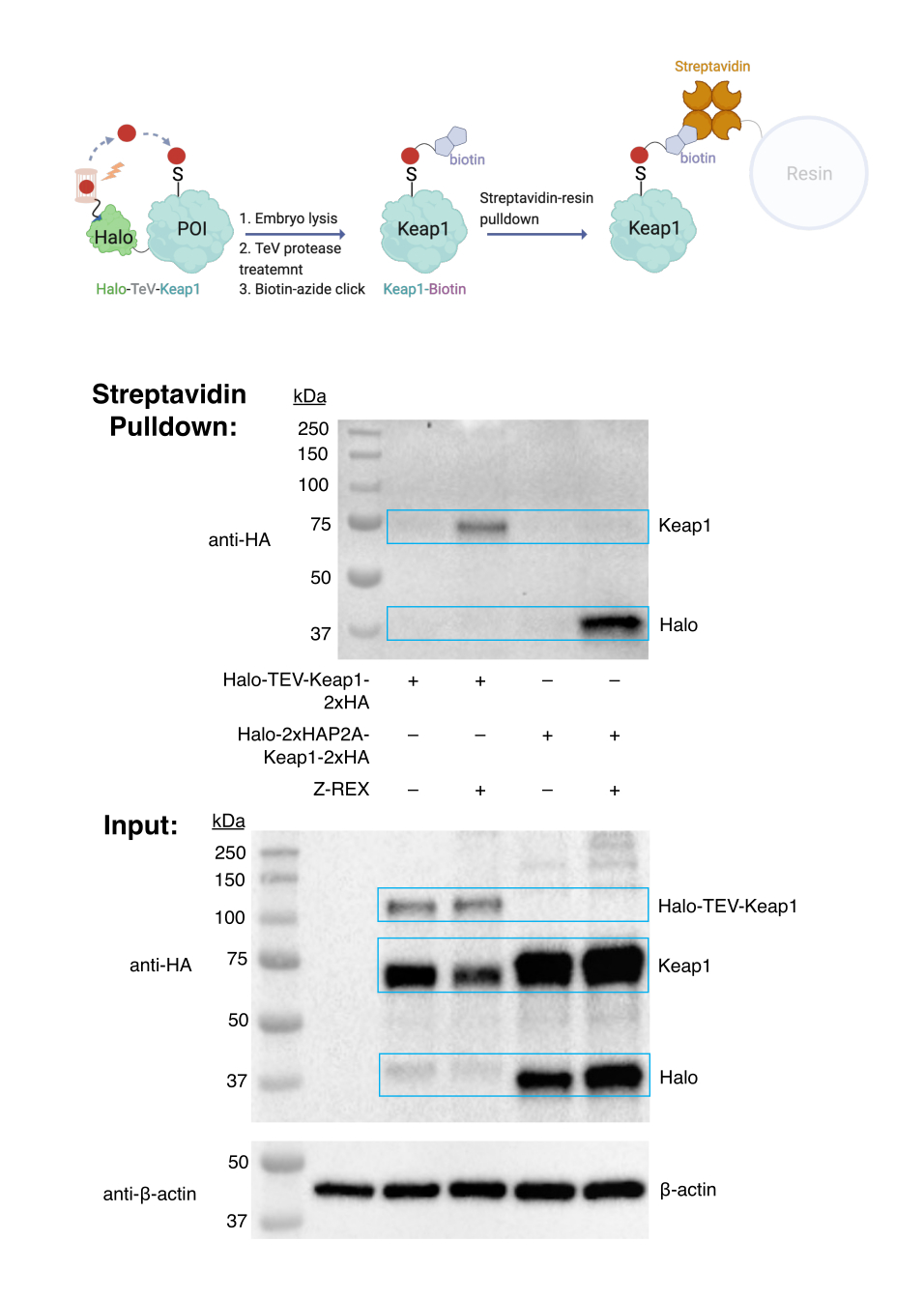
Biotin azide-click coupling and biotin pull-down assay. Target-labeling assessment. (See also Figure 3). The target-labeling assessment was carried out using WT embryos, injected with mRNA encoding either Halo-TeV-Keap1-2xHA (Halo-POI fusion construct) or Halo-2xHA-P2A-Keap1-2xHA (P2A-split construct, in which Halo and Keap1 are not fused; Figure 1D). Labeled Keap1 protein was only pulled down in the group expressing fusion protein and treated with Z-REX (second lane in the top anti-HA blot), but not in other control groups (no mRNA injection, fusion construct without Z-REX, or P2A-split construct). The results indicate the HNE(alkyne) was successfully delivered to Keap1, and the modified Keap1 was subsequently conjugated with biotin through click reaction, and the biotin-labeled Keap1 was pulled down by streptavidin resin.
Transcriptional analysis. RNA-seq and qRT-PCR. (See also Figure 4). The transcriptional change after Z-REX treatment was assessed by RNA-seq and qRT-PCR. In RNA-seq, several immune-related genes were downregulated after Z-REX. In contrast, many antioxidant response(AR)-related genes were upregulated after Z-REX, which resulted from the induction of the Keap1-Nrf2-AR pathway upon HNEylation on Keap110 (Figure 4A). In qRT-PCR analysis, similar results were found when analyzing three immune-related genes (lyz, mpeg1.1, and coro1a) (Figure 4B). The up- and down-regulation of the respective genes showed the successful induction of pathways mediated by Keap1 HNEylation.
Whole-mount (co-)immunofluorescence staining assay and colocalization analysis. (See also Figure 5). The exogenous Halo-TeV-Keap1-2xHA and Halo-2xHA-P2A-Keap1-2xHA expression were assessed by whole-mount immunofluorescence (IF) staining (Figure 5A,B). The P2A-split-construct had two times the number of HA tags than the TeV-fusion-construct, which corresponds to a twofold higher anti-HA signal in the P2A-split-construct-mRNA-injected group than the other, indicating the expression level of the two constructs were similar (Figure 5C). The expression levels of the Halo-TeV-Keap1 (wt) and Halo-TeV-Keap1(C151S,C273W,C288E) were also found similar when probing with anti-Halo (Figure 5D,E). Colocalization of neutrophils and active caspase 3 in Z-REX-treated Tg(lyz:TagRFP) was observed by co-immunostaining with anti-RFP and anti-active-Caspase 3 (Figure 5F). Active Caspase 3 is an indicator of apoptosis events.

Figure 1: Z-REX workflow. (A,B) A 1-4 cell stage zebrafish embryo is injected with (morpholino and) mRNA encoding Halo-POI (e.g., Halo-Keap1). Injected embryos are then treated with a probe composed of a HaloTag ligand and a photocaged electrophile appended with an alkyne functional group, such as Ht-PreHNE(alkyne) in B. After removing the excess amount of probe, the embryo is exposed to light to release the electrophile of interest [e.g., HNE or its analog, HNE(alkyne)]. The downstream analysis is performed at a given/user-defined timepoint. (C) Design and mechanism of the Ht-PreLDE probe, which is applicable to different lipid-derived electrophiles (LDE). (D) Negative/technical control groups for Z-REX. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Live imaging of transgenic neutrophil/macrophage reporter fish subjected to Z-REX. Z-REX-mediated Keap1 HNEylation induces neutrophil/macrophage apoptosis. (A) Representative images of Tg(lyz:TagRFP) fish expressing either Halo-TeV-Keap1 (fusion construct) or Halo-P2A-Keap1 (split construct), and subjected to negative control conditions [no treatment, light alone, or Ht-PreHNE(alkyne) alone or Z-REX]. Embryo age: 36 hpf. (B) Quantitation of neutrophil levels in A. (C) Representative images of Tg(mpeg1:eGFP) fish expressing Halo-TeV-Keap1 with or without Z-REX treatment. Embryo age: 34 hpf. (D) Quantitation of macrophage levels in C. (E,F) Time-course measurement of (E) neutrophil and (F) macrophage levels after Z-REX treatment. (G) Similar experiment as in A in fish expressing either Halo-TeV-Keap1 (WT) or Halo-TeV-Keap1 (C151S, C273W, C288E), a mutant that does not have HNE-sensing capability. (H) Quantitation of neutrophil levels in G. Scale bars: 500 µm. All the graphs are presented with mean ± SEM. p values were calculated with one-way ANOVA (blue) and two-tailed Student's t-test (black). This figure has been modified from Poganik et al.7. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Biotin pull-down assay. WT embryos expressing Halo-TeV-Keap1-2XHA or Halo-2XHA-P2A-Keap1-2XHA were treated with Z-REX or respective negative control conditions (no probe treatment in this case). After harvest, embryos were lysed and treated with TeV protease before the biotin pull-down assay. The results were analyzed by western blotting. This figure has been modified from Huang et al. Z-REX: shepherding reactive electrophiles to specific proteins expressed either tissue-specifically or ubiquitously, and recording the resultant functional electrophile-induced redox responses in larval fish. This figure has been modified from Huang et al.11. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Transcriptional analysis. (A) RNA-seq results of Z-REX-treated versus non-treated embryos. Statistically significant differentially-expressed (SDE) genes are highlighted. Immunity-related SDE genes are colored red. Antioxidant response (AR)-related genes are colored green. Other SDE genes are colored blue. All p values were calculated with CuffDiff. (B-D) Three immunity-related SDE genes from A: (B) lyz, (C) mpeg1.1, and (D) coro1a were further analyzed with qRT-PCR, and only the Z-REX-treated embryos showed the suppression of these transcripts. All the graphs are presented with mean ± SEM. p values were calculated with one-way ANOVA (blue) and two-tailed Student's t-test (black). This figure has been modified from Poganik et al.7. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Whole-mount immunofluorescence staining assay. (A,B) Representative images of embryos expressing either (A) Halo-TeV-Keap1-2xHA or (B) Halo-2xHA-P2A-Keap1-2xHA immunostained with anti-HA and secondary antibody conjugated with AlexaFluor568. mRNA-injected fish were compared to age-matched non-injected fish. (C) Quantification of anti-HA signal in (A,B). (D) Representative images of embryos expressing Halo-TeV-Keap1 (WT) or Halo-TeV-Keap1 (C151S, C273W, C288E) immunostained with anti-Halo and secondary antibody conjugated with AlexaFluor647. mRNA-injected fish were compared to age-matched non-injected fish. (E) Quantification of anti-Halo signal in D. p values were calculated with two-tailed Student's t-test. (F) Tg(lyz:TagRFP) embryos subjected to Z-REX were co-immunostained with anti-RFP and anti-active Caspase 3, and respective fluorophore-conjugated secondary antibodies. The white box marks the magnified area. White arrows indicate colocalizations of neutrophils and active Caspase 3. Scale bars: 500 µm. All the graphs are presented with mean ± SEM. This figure has been modified from Poganik et al.7. and Huang et al.11. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Table 1: List of buffers used in this study. Please click here to download this File.
Dyskusje
Z-REX described in this protocol demonstrates a robust strategy for electrophile-target pair investigation and signaling pathway deconvolution in live fish. The proximity-directed delivery enables dosage and spatial control of the electrophilic compound treatment. Unlike conventional bolus dosing methods, in which the supraphysiological concentrations of electrophile deployed often lead to off-target issues, the relatively minor amount of electrophile released to the system renders Z-REX largely noninvasive. We have used 0.1-6 µM Ht-PreHNE(alkyne) in zebrafish embryos, and the results showed the treatment is not detrimental to embryo development11.
The Z-REX procedure is generally longer than T-REX, a technique for screening/studying electrophile-sensing proteins in cultured cells. Suppose the experiment's purpose is to screen for electrophile-target interactions; we suggest first performing extensive screening by T-REX in cultured cells and using Z-REX for in vivo validation and phenotypic/pathway analysis. Compared to cell culture, requirements for performing Z-REX are basic fish husbandry techniques in addition to biochemical experimental skills required by T-REX. A typical timeframe for Z-REX (from fish crossing to light-inducible electrophile delivery) is 2-3 days, which is no more than 1 day longer than the typical time for a T-REX experiment on transfected live cells. Live imaging for phenotypic analysis can be performed 2-10 h after light illumination; click coupling with biotin-azide for pull-down assay takes 3 days; qRT-PCR for assaying transcriptional response takes 3 days; IF staining takes 5 days. These steps are roughly similar to their cell culture equivalents, although the interpretation of data requires an understanding of fish physiology and reporter strains.
As a multiple variable procedure12, several control groups are necessary for Z-REX to exclude uncertainties in the results (Figure 1D). Common control groups are: (1) DMSO/vehicle treatment only; (2) probe treatment, but without light illumination; (3) light illumination, but without probe treatment; (4) P2A-split construct, in which Halo and the POI are expressed separately, so the proximity delivery is ablated; and (5) hypomorphic mutants, whose electrophile-sensing residue(s) is/are mutated, such as Akt3 (C119S)6 and Keap1 (C151S, C273W, C288E)5, which we have used in previous studies.
If downstream assays involve western blot analysis, deyolking must be performed before harvest. The yolk proteins reduce the fidelity of lysate-concentration assessments and may bind non-specifically to antibodies. When performing live fish imaging or whole-mount IF staining, we have also observed non-specific fluorescent signals in the yolk sac, likely resulting from autofluorescent proteins in the yolk sac, or non-specific binding of the antibodies themselves. If the autofluorescence signal interferes with the signal, we suggest excluding the yolk sac from quantification, or quantifying different regions separately. Dechorination is necessary for live fish imaging and whole-mount IF staining assay. The chorion can interfere with imaging, and later with quantification/cell counting. However, dechorination is only applicable to embryos older than 1 dpf; younger embryos at blastulation/gastrulation/segmentation stages are too fragile to be dechorionated.
The Z-REX protocol described here is based on mRNA-driven ectopic POI expression. The procedure is rapid compared to using/generating transgenic fish lines. mRNA-driven expression is ubiquitous and transient, and lasts for at least 2 days for mRNAs used in this protocol. However, the duration of expression is likely to vary in other cases. Thus, this approach provides a fast, and more global investigative window into the effects of a specific electrophile-labeling event, compatible with several high-throughput/high-content assays. Transgenic lines with stable Halo-POI expression in specific tissues are also compatible with Z-REX11. Such lines are best used when a more precise question needs to be asked, for instance, when a phenotype in a specific organ is predicted from cell-culture data, or when screening from mRNA-injection experiments predicts that a specific organ is sensitive to an electrophile-labeling event. A heart-specific antioxidant response induction through Z-REX was demonstrated using Tg(gstp1:GFP;DsRed-P2A-myl7:Halo-TeV-Keap1) fish in our previous publication11. It may also be possible to perform Z-REX on transgenic fish older than 2 dpf.
Ujawnienia
Isoform-specific small-molecule kinase inhibitors, the discovery of which were enabled by REX technologies, have been filed for patent application
Podziękowania
Funding: Novartis FreeNovation, NCCR, and EPFL.
Materiały
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol (BME) | Sigma-Aldrich | M6250 | |
| 1.5-mL microcentrifuge tube | Axygen | MCT-150-C-S | |
| 10-cm Petri dishes | Celltreat | 229692 | |
| 2-mL microcentrifuge tube | Axygen | MCT-200-C-S | |
| 30% Acrylamide and bis-acrylamide solution | BioRad | 1610154 | |
| 6-well plate | Celltreat | 229506 | |
| Acetone | Fisher | A/0600/15 | |
| Agarose | GoldBio | A201-100 | |
| All Blue Prestained Protein Standards | BioRad | 1610373 | |
| Ammonium persulfate | Sigma | A3678 | |
| Biotin-dPEG11-azide | Quanta Biodesign | 102856-142 | |
| Bradford Dye | BioRad | 5000205 | |
| BSA | Fisher | BP1600-100 | |
| Capillary tubes | VWR | HARV30-00200 | |
| Chloroform | Supelco, Inc | 1.02445.1000 | |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 | |
| Cu(TBTA) | Lumiprobe | 21050 | |
| CuSO4 | Sigma | 209198 | |
| DMSO | Fisher | D128-500 | |
| Donkey anti-mouse-Alexa Fluor 647 | Abcam | ab150107 | 1:800 (IF) |
| Donkey anti-rabbit-Alexa Fluor 647 | Abcam | ab150075 | 1:1000 (IF) |
| Donkey anti-rat-AlexaFluor 568 | Abcam | ab175475 | 1:1000 (IF) |
| ECL substrate | Thermo Fisher Scientific | 32106 | |
| ECL-Plus substrate | Thermo Fisher Scientific | 32132 | |
| End-to-end rotator | FinePCR | Rotator AG | |
| Ethanol | Fisher | E/0650DF/15 | |
| Ethidium bromide | Sigma | E1510 | |
| Flaming/Brown Micropipette Puller | Sutter Instrument Co. | P-97 | |
| Fluorescence stereomicroscope | Leica | M165 FC | |
| Forceps (blunt) | self made | self made by blunting sharp forceps (Fine Science Tools, Dumont #5, 11252-40) | |
| Forceps (sharp) | Fine Science Tools | Dumont #5, 11252-40 | |
| Gel loading tip | Fisher | 02-707-181 | |
| Gel/blot imager | Vilber | Fusion FX imager | |
| Glass beads | Sigma | 45-G1145 | |
| Glass stage micrometer | Meiji Techno. | MA285 | |
| Heat inactivated FBS | Sigma | F2442 | |
| Heated ultrasonic bath | VWR | 89375-470 | |
| HEPES | Fisher | BP310-1 | |
| High capacity streptavidin agarose | Thermo Fisher Scientific | 20359 | |
| Ht-PreHNE alkyne probe | self-made | - | Parvez, S. et al. T-REX on-demand redox targeting in live cells. Nat Protoc. 11 (12), 2328-2356, (2016). |
| Imaging plate (10% HBSS buffer, for live embryos) | Made with Petri dish, and 2% agarose in 10% HBSS buffer | ||
| Imaging plate (PBS, for formaldehyde-fixed embryos) | Made with Petri dish, and 2% agarose in PBS | ||
| Injection plate | Made with microinjection mold, Petri dish, and 2% agarose in 10% HBSS buffer | ||
| LDS | Apollo | APOBI3331 | |
| Methanol | VWR | 20864.32 | |
| Microinjection mold | Adaptive Science Tools | TU-1 | |
| Microloader tips | Eppendorf | 930001007 | |
| Micromanipulator | Narishige | MN-153 | |
| Microscope for micro-injection | Olympus | SZ61 | |
| Microscope light source | Olympus | KL 1600 LED | |
| Mineral oil | Sigma | M3516 | |
| mMessage mMachine SP6 Transcription Kit | Ambion | AM1340 | |
| Mouse anti- β-actin-HRP | Sigma | A3854 | 1:20000 (WB) |
| Mouse anti-HaloTag | Promega | G921A | 1:500 (IF) |
| Multi-mode reader | BioTek Instruments | Cytation 5 | |
| Nucleic acid agarose gel electrophoresis apparatus | Biorad | Mini-Sub Cell GT Systems | |
| Oligo(dT)20 | Integrated DNA Technologies | customized: (dT)20 | |
| Orange G | Sigma | O3756 | |
| Paraformaldehyde | Sigma | P6148 | |
| PBS | Gibco | 14190144 | |
| pCS2+8 Halo-2XHA-P2A-TeV-Keap1-2xHA | self-cloned | - | Available from Prof. Yimon AYE's group at EPFL |
| pCS2+8 Halo-TeV-Keap1-2xHA | self-cloned | - | Available from Prof. Yimon AYE's group at EPFL |
| Pneumatic PicoPump | WPI | SYS-PV820 | |
| Protein electrophoresis equipment and supplies | Biorad | Mini-PROTEAN Tetra Vertical Electrophoresis | |
| Rabbit anti-active Caspase-3 | BD Pharmingen | 559565 | 1:800 (IF) |
| Rat anti-HA-HRP | Sigma | H3663 | 1:500 (IF and WB) |
| Rat anti-RFP | ChromoTek | 5F8 | 1:800 (IF) |
| Refrigerated centrifuge | Eppendorf | 5417R | |
| RNAseOUT recombinant ribonuclease inhibitor | ThermoFisher Scientific | 10777019 | |
| RnaseZap RNA decontamination solution | ThermoFisher Scientific | AM9780 | |
| Rocking Shaker | DLAB | SK-R1807-S | |
| SDS | Teknova | S9974 | |
| SuperScript III reverse transcriptase | ThermoFisher Scientific | 18080085 | |
| t-Butanol | Sigma | 471712 | |
| TCEP-HCl | Gold Biotechnology | TCEP1 | |
| TeV protease (S219V) | self-made | - | Parvez, S. et al. T-REX on-demand redox targeting in live cells. Nat Protoc. 11 (12), 2328-2356, (2016). |
| Tg(lyz:TagRFP) | Zebrafish International Resource Center (ZIRC) | uwm11Tg (ZFIN) | - |
| Tg(mpeg1:eGFP) | Zebrafish International Resource Center (ZIRC) | gl22Tg (ZFIN) | - |
| Thermal cycler | Analytik Jana | 846-x-070-280 | |
| TMEDA | Sigma | T7024 | |
| Transfer pipets | Fisher | 13-711-9D | |
| Tris | Apollo | APOBI2888 | |
| Triton X-100 | Fisher | BP151-100 | |
| TRIzol reagent | Thermo Fisher Scientific | 15596018 | |
| Trypsin inhibitor from Glycine max (soybean) | Sigma | T9003 | |
| Tween 20 | Fisher | BP337-500 | |
| UV lamp with 365-nm light | Spectroline | ENF 240C | |
| UV meter | Spectroline | XS-365 | |
| Vortexer | Scientific Industries, Inc. | Vortex-Genie 2 | |
| Western Blotting Transfer equipment and supplies | Biorad | Mini Trans-Blot or Criterion Blotter | |
| Zebrafish husbandry and breeding equipment | in house | ||
| Zirconia beads | BioSpec | 11079107zx |
Odniesienia
- Long, M. J. C., Ly, P., Aye, Y. A primer on harnessing non-enzymatic post-translational modifications for drug design. RSC Medicinal Chemistry. 12 (11), 1797-1807 (2021).
- Parvez, S., Long, M. J. C., Poganik, J. R., Aye, Y. Redox signaling by reactive electrophiles and oxidants. Chemical Reviews. 118 (18), 8798-8888 (2018).
- Parvez, S., et al. T-REX on-demand redox targeting in live cells. Nature Protocols. 11 (12), 2328-2356 (2016).
- Long, M. J. C., et al. Precision electrophile tagging in Caenorhabditis elegans. Biochemistry. 57 (2), 216-220 (2018).
- Van Hall-Beauvais, A., Zhao, Y., Urul, D. A., Long, M. J. C., Aye, Y. Single-protein-specific redox targeting in live mammalian cells and C. elegans. Current Protocols in Chemical Biology. 10 (3), 43 (2018).
- Long, M. J. C., et al. Akt3 is a privileged first responder in isozyme-specific electrophile response. Nature Chemical Biology. 13 (3), 333-338 (2017).
- Poganik, J. R., et al. Wdr1 and cofilin are necessary mediators of immune-cell-specific apoptosis triggered by Tecfidera. Nature Communications. 12 (1), 5736 (2021).
- Lin, H. -. Y., Haegele, J. A., Disare, M. T., Lin, Q., Aye, Y. A generalizable platform for interrogating target- and signal-specific consequences of electrophilic modifications in redox-dependent cell signaling. Journal of the American Chemical Society. 137 (19), 6232-6244 (2015).
- Schmittgen, T. D., Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nature Protocols. 3 (6), 1101-1108 (2008).
- Wakabayashi, N., et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nature Genetics. 35 (3), 238-245 (2003).
- Huang, K. -. T., et al. Z-REX: Shepherding reactive electrophiles to specific proteins expressed either tissue-specifically or ubiquitously, and recording the resultant functional electrophile-induced redox responses in larval fish. Nature Protocols. , (2023).
- Long, M. J. C., Assari, M., Aye, Y. Hiding in plain sight: The issue of hidden variables. ACS Chemical Biology. 17 (6), 1285-1292 (2022).
Przedruki i uprawnienia
Zapytaj o uprawnienia na użycie tekstu lub obrazów z tego artykułu JoVE
Zapytaj o uprawnieniaThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Wszelkie prawa zastrzeżone