
Method Article
Analyses of Actin Dynamics, Clutch Coupling and Traction Force for Growth Cone Advance
In This Article
Summary
To advance, growth cones must exert traction forces against the external environment. The generation of traction forces is dependent on actin dynamics and clutch coupling. The present study describes methods for analyzing actin dynamics, clutch coupling and traction forces for growth cone advance.
Abstract
To establish functional networks, neurons must migrate to their appropriate destinations and then extend axons toward their target cells. These processes depend on the advances of growth cones that located at the tips of neurites. Axonal growth cones generate driving forces by sensing their local microenvironment and modulating cytoskeletal dynamics and actin-adhesion coupling (clutch coupling). Decades of research have led to the identification of guidance molecules, their receptors, and downstream signaling cascades for regulating neuronal migration and axonal guidance; however, the molecular machineries required for generating forces to drive growth cone advance and navigation are just beginning to be elucidated. At the leading edge of neuronal growth cones, actin filaments undergo retrograde flow, which is powered by actin polymerization and actomyosin contraction. A clutch coupling between F-actin retrograde flow and adhesive substrate generates traction forces for growth cone advance. The present study describes a detailed protocol for monitoring F-actin retrograde flow by single speckle imaging. Importantly, when combined with an F-actin marker Lifeact, this technique can quantify 1) the F-actin polymerization rate and 2) the clutch coupling efficiency between F-actin retrograde flow and the adhesive substrate. Both are critical variables for generating forces for growth cone advance and navigation. In addition, the present study describes a detailed protocol of traction force microscopy, which can quantify 3) traction force generated by growth cones. Thus, by coupling the analyses of single speckle imaging and traction force microscopy, investigators can monitor the molecular mechanics underlying growth cone advance and navigation.
Introduction
In the developing vertebrate brain, neurons undergo elaborately organized migrations and project axons toward appropriate synaptic partners to establish functional neuronal networks1,2,3. Growth cones, which are sensory and motile structures located at the tip of neurites, determine the speed and direction of neuronal migration and axon outgrowth3,4,5. Since neurons are surrounded by tightly packed environments, growth cones must exert forces against their environment to move forward6,7. To understand the mechanisms underlying neuronal migration and axonal guidance, analyses of the molecular mechanics for growth cone advance are essential.
Decades of analysis have revealed that traction force to drive growth cone advance is generated by the 'clutch' mechanism; this mechanism is thought to function not only in the axonal growth cone but also in the leading process growth cone of migrating neurons8,9,10,11,12. Namely, actin filaments (F-actins) in growth cones polymerize at the leading edge and depolymerize proximally, pushing out the leading-edge membrane13,14,15. The resultant force, in conjunction with actomyosin contraction, induces rearward movement of F-actins called retrograde flow7,11,16,17,18,19,20,21. Clutch- and cell adhesion molecules mediate mechanical coupling between F-actin retrograde flow and the adhesive substrate and transmit the force of F-actin flow onto the substrate, thereby generating traction force for growth cone advance7,8,9,11,12,22. Concurrently, the actin-substrate coupling reduces the F-actin flow velocity and converts actin polymerization into the force to protrude the leading-edge membrane9,10.
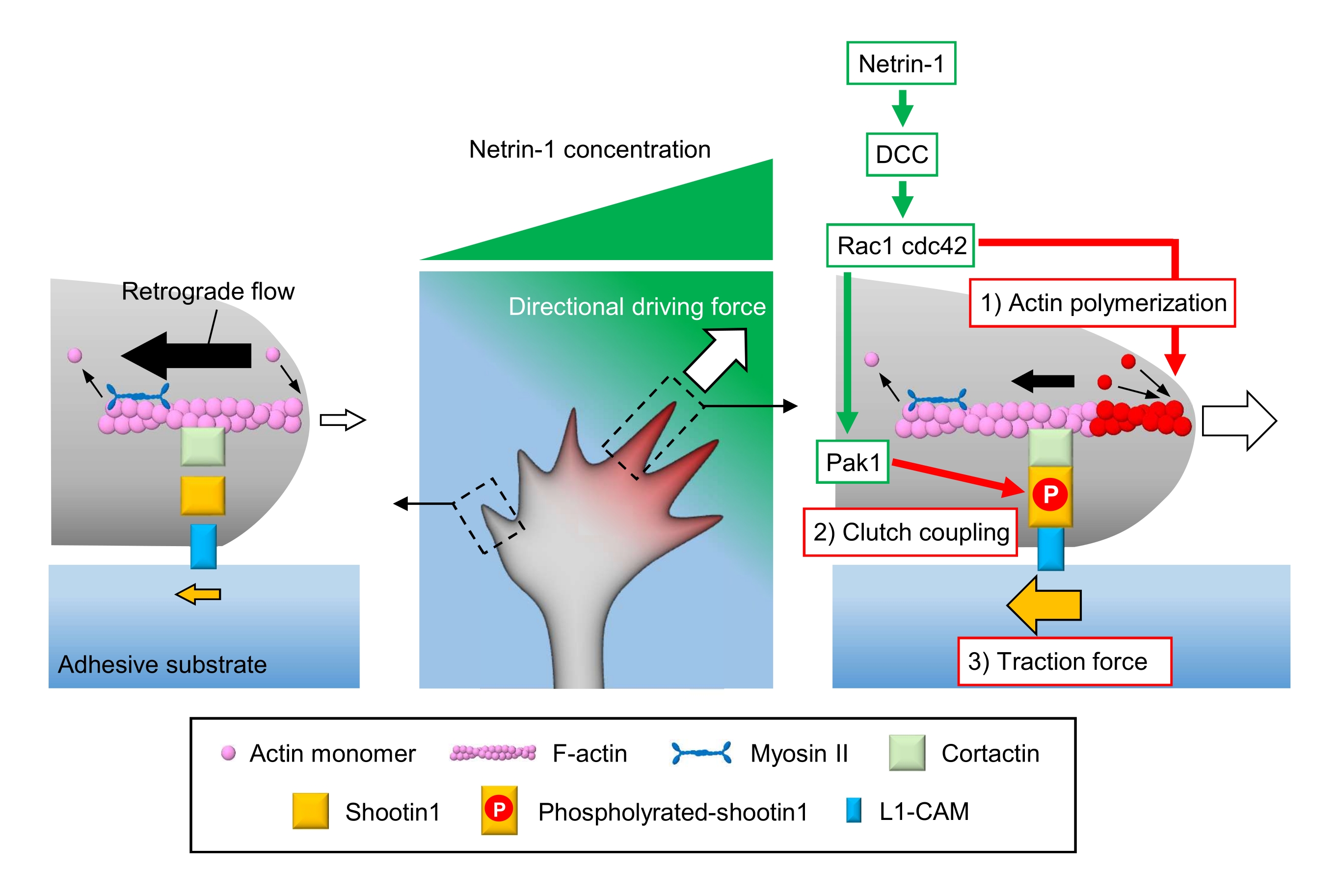
Axonal growth cones sense local chemical cues and transduce them into a directional driving force for growth cone navigation3,23,24,25. For example, an axon guidance molecule netrin-1 stimulates its receptor deleted in colorectal cancer (DCC), and activates the Rho guanosine triphosphate (GTP)-binding proteins cell division control protein 42 (Cdc42) and Ras-related C3 botulinum toxin substrate 1 (Rac1), and their downstream kinase p21-activated kinase 1 (Pak1)26. Cdc42 and Rac1 promote 1) actin polymerization, and Pak1 phosphorylates a clutch molecule shootin122,26. Shootin1 interacts with F-actin retrograde flow via an actin-binding protein cortactin27. Shootin1 also interacts with L1 cell adhesion molecule (L1-CAM)20,24. Shootin1 phosphorylation increases the binding affinities for cortactin and L1-CAM, and enhances shootin1-mediated 2) clutch coupling24,27. Within the growth cone, asymmetrical activations of actin polymerization and clutch coupling increase 3) traction force on the side of the netrin-1 source, thereby generating directional driving force for growth cone turning (Figure 1)24. Intensive research over the last few decades with respect to neuronal migration and axon guidance has enhanced the understanding of guidance molecules, their receptors, and associated downstream signaling cascades2,10,28,29,30. However, the molecular machineries to generate forces for growth cone advance are just beginning to be elucidated; this may be attributed to the limited usage of the protocols for mechanobiological analyses.
The present study describes a detailed protocol for monitoring F-actin retrograde flow by single speckle imaging16,18. Monitoring of F-actin retrograde flow has been extensively performed using super-resolution microscopy, spinning-disk confocal microscopy and total interference reflection fluorescence (TIRF) microscopy25,31,32,33,34,35,36,37,38. The protocol in the present study, however, uses a standard epifluorescence microscope and is thus readily adoptable11,16,18,20,22,23,24,27,39,40,41,42. When combined with F-actin labelling by Lifeact43, single speckle imaging allows for quantifications of the actin polymerization rate and the clutch coupling efficiency between F-actin retrograde flow and the adhesive substrate39,42. The present study further describes a detailed protocol of traction force microscopy using a fluorescent bead-embedded polyacrylamide (PAA) gel11,22,23,24,27,39,41,42,44. This method detects and quantifies traction force under the growth cone by monitoring force-induced bead movements44,45. An open-source traction force analysis code is provided, and the method for quantifying traction force during growth cone migration is explained in detail. With the aid of single speckle imaging and traction force microscopy, understanding the molecular mechanics underlying growth cone migration and navigation will be facilitated. These techniques are also applicable for analyzing the molecular mechanics underlying dendritic spine enlargement, which is known to be important in learning and memory42.
Protocol
All experiments using laboratory animals were performed with the Institutional Animal Care and Use Committee of Nara Institute of Science and Technology. Investigators should follow established guidelines by their institutional and national animal regulatory committees for the care and use of laboratory animals.
1. Preparation of solutions and media
- Prepare the solutions and culture media as summarized in Supplemental File 1
2. Preparation of poly-D-lysine (PDL)/laminin-coated substrates
- Coat 14 mm-diameter glass-bottom dishes with 100 µg/mL PDL, dissolved in phosphate-buffered saline (PBS, pH 7.4), and incubate for overnight in a humidified incubator at 37 °C.
NOTE: Do not dry up the glass surfaces after this step - Remove the PDL solution and pipette 1 mL of PBS on the glass for 10 s three times.
- Coat the dishes with 5 µg/mL laminin, dissolved in PBS, and incubate for overnight in a humidified incubator at 37 °C.
- Remove the laminin solution and pipette 1 mL of PBS on the glass for 10 s three times.
- Remove the PBS and place 0.5 mL of the neurobasal medium on the glass surfaces. Store the dishes in a humidified incubator at 37 °C.
NOTE: PDL/laminin-coated dishes can be stored for 2-3 days in a humidified incubator at 37 °C.
3. Dissection and dissociation of the hippocampus
- Euthanize a pregnant mouse by cervical dislocation.
NOTE: The present study uses commercially available mice (see Table of Materials). Investigators should use mice that are bred and humanely treated in a sterilized environment. - Dissect out embryonic day-16 (E16) mouse embryos, and place them on ice.
- In a laminar flow hood, dissect out the brains with scissors and place them on a sterile 10 cm dish containing 10 mL of ice-cold dissection solution.
- With a dissection stereo microscope, carefully peel away the meninges of the cerebral hemispheres and then dissect out the hippocampi using forceps.
- Transfer the hippocampi into 5 mL of ice-cold digestion solution in a 15 mL tube. Incubate the hippocampi in a water bath for 20 min at 37 °C.
- Remove the digestion solution and add 3 mL of dissection solution.
- Gently pipette the hippocampi four times with a Pasteur pipette.
- Incubate the hippocampi in a water bath for 20 min at 37 °C.
- Transfer the cell suspension into a 50 mL centrifuge tube. Add 3 mL of new dissection solution to the undissociated tissue.
- Repeat Steps 3.7-3.9 until the hippocampi are completely dissociated.
- Remove the floating aggregates of DNA from the cell suspension by swirling and aspirating with a Pasteur pipette. Centrifuge the cell suspension at 180 x g for 20 min at 4 °C.
NOTE: DNA derived from damaged cells will disturb the centrifugation process. If neurons remain in the supernatant, centrifuge the cell suspension again after carefully removing the DNA. - Equilibrate neurobasal medium containing 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 µg/mL) in a humidified incubator at 37 °C with 5% CO2.
4. Transfection and culturing neurons
- Remove the supernatant and add 5 mL of ice-cold PBS into the 50 mL tube. Resuspend the cell pellet by gentle pipetting.
- Transfer 10 µL of the cell suspension into a microcentrifuge tube, add 10 µL of 0.5% trypan blue solution, and then count the number of cells with a hemocytometer.
- Centrifuge the cell suspension in the 50 mL tube at 180 x g for 20 min at 4 °C.
- In the meantime, aliquot for transfection, 1 µg of pFN21A-HaloTag-actin and 3 µg of pmNeonGreen-N1-Lifeact per 1 x 106 cells, into a microcentrifuge tube.
- Following centrifugation, remove the supernatant and add 100 µL of electroporation medium (provided by transfection kit) into the 50 mL tube. Resuspend the cell pellet by pipetting.
- Mix the cell suspension with the aliquoted DNA solution and transfer the mixture to a cuvette (provided by the transfection kit).
NOTE: Since air bubbles disturb electroporation, remove them from the cell suspension. - Insert the cuvette into the electroporation apparatus (Table of Materials), and perform electroporation using program O-005.
- Immediately add 1 mL of the pre-warmed and equilibrated neurobasal medium containing 10% FBS, penicillin (100 U/mL), and streptomycin (100 µg/mL) to the cuvette.
- Transfer the cell suspension to a 15 mL centrifuge tube using a plastic pipette (provided by the transfection kit).
- Transfer 10 µL of the cell suspension into a microtube, add 10 µL of 0.5% trypan blue solution, and then count the number of cells with a hemocytometer.
- Take out PDL/laminin-coated glass-bottom dishes from the incubator and remove the neurobasal medium.
- Pipette 0.5 mL of the cell suspension containing 2.0 x 105 cells per dish and incubate in a humidified incubator at 37 °C with 5% CO2 for 3 h.
- Replace the medium with 0.5 mL of neurobasal medium containing 2% B-27 supplement, glutamine (1 mM), penicillin (100 U/mL), and streptomycin (100 µg/mL). Culture the neurons in a humidified incubator for 3 days at 37 °C with 5% CO2.
5. Single speckle imaging at neuronal growth cones
- On day in vitro (DIV) 3, treat the neurons with tetramethyl-rhodamine (TMR) ligand at a dilution of 1:2000 dilution in culture medium. Maintain the neurons for 1 h at 37 °C with 5% CO2.
- Wash the TMR ligand three times with pre-warmed PBS.
- Remove the PBS and pipette 0.5 mL of warmed Leibovitz's L-15 medium containing 2% B-27 supplement, glutamine (1 mM), penicillin (100 U/mL), and streptomycin (100 µg/mL).
- Maintain neurons for 1 h at 37 °C.
- Turn on an epifluorescence microscope and set the stage-top incubator to 37 °C.
NOTE: The present protocol uses an epifluorescence microscope equipped with a complementary metal-oxide semiconductor camera, a 100x/1.40 NA oil immersion objective lens, a mercury lamp and an image acquisition software (see Table of Materials). Other epifluorescence microscopes with equivalent specifications may also be used for this analysis. - Place the TMR ligand-treated neurons in the glass-bottom dish on the warmed stage-top incubator.
- Set the image acquisition parameters as follows: exposure time, 500 ms for Lifeact and HaloTag-actin fluorescent channels; binning, 1 x 1 (0.065 µm × 0.065 µm per pixel); time interval, 3 s; duration, 50 frames.
- Select a growth cone that strongly expresses Lifeact and weakly expresses HaloTag-actin.
NOTE: Lifeact expression should be strong enough to visualize growth cone morphology. HaloTag-actin expression should be weak insufficient to be detected (Figure 2A). The levels of fluorescent protein expressions can be adjusted by varying the amount of DNA for transfection. - Close the field of the diaphragm to illuminate a minimum area that includes the growth cone (Figure 2B).
NOTE: The illumination of a minimum area reduces the background signal and increases the fluorescence signal to noise (S/N) ratio, enabling the detection of actin speckles (Figure 2B). To further increase the S/N ratio, illuminating the growth cone without reducing the light by neutral density filters is recommended. - Acquire time-lapse images (Supplemental File 2). Save as a multichannel time-lapse stack image (tiff format).
6. Quantifications of F-actin flow velocity and polymerization rate using an image processing and analysis software Fiji
NOTE: Refer to Supplemental File 3 for practice data for quantifying F-actin flow velocity and the actin polymerization rate.
- Open the multichannel time-lapse stack image on Fiji.
- Select Analyze > Set Scale, and set the pixel size of the images.
- Find an actin speckle that flows retrogradely for a minimum of five time-frames within an F-actin bundle in filopodia or lamellipodia.
- Select Image > Transform > Rotate, and adjust the angle of the images so that the F-actin bundle points upward (Figure 3A).
- Click on Rectangle on the toolbar, and draw a box that includes actin speckles and the tip of the F-actin bundle (Figure 3B, 1 and 2).
NOTE: The signals of HaloTag-actin speckles are weak. In addition, Lifeact signals at the distal tips of the growth cone are weak and faint (Figure 2B) because the distal tips of the growth cone is thin compared to the proximal part. Investigators should enhance the signals to optimize the quantifications of F-actin flow velocity and polymerization rate (Figure 3B). Although the Lifeact signal at the proximal growth cone becomes saturated, this will not disturb the determination of the distal end of F-actins (Figure 3E, yellow line). - Select Image > Duplicate and input the five time-frames (Figure 3B, 3-5).
- Select Image > Stacks > Make Montage, and input the parameters: Columns, 5; Rows, 1; Scale factor, 1.
- Click on OK. The image montage will appear on the screen.
- Select Image > Color > Split Channels. This separates the two fluorescent channels.
- Quantification of F-actin flow velocity.
- Select Analyze > Set Measurements, and choose Area and Bounding Rectangle.
- Click on Oval on the toolbar, and draw a circle on an actin speckle (Figure 3C, 1 and 2).
- Select Image > Overlay > Add Selection. The circle is overlaid on the image montage (Figure 3C, 3, and 4).
NOTE: The color of the overlay can be changed at Edit > Options > Colors. - Repeat the overlaying of circles for the remaining speckles (Figure 3C, 5).
NOTE: To accurately determine the centers of actin speckle, investigators should overlay circles on the speckles. - Click on Straight on the toolbar, and draw a line that links the centers of the circles (Figure 3D, 1 and 2).
- Select Analyze > Measure. The result will appear on the screen (Figure 3D, 3).
NOTE: Height displayed in the result indicates the translocation distance of the actin speckle during the observation (Figure 3D, 3). - Calculate the F-actin flow velocity by dividing the translocation distance by the observation time.
- Quantification of the F-actin polymerization rate.
- Draw a line that links the tips of the F-actin bundle (Figure 3E, 1).
- Select Analyze > Measure. The result will appear on the screen (Figure 3E, 2).
NOTE: Height in the result indicates the extension length of the F-actin bundle during the observation (Figure 3E, 2). - Calculate the F-actin extension rate by dividing the extension length by the observation time.
- Calculate the F-actin polymerization rate as the sum of the F-actin flow velocity and the extension rate (Figure 3F).
7. Preparation of a PAA gel for traction force microscopy and neuron cultures
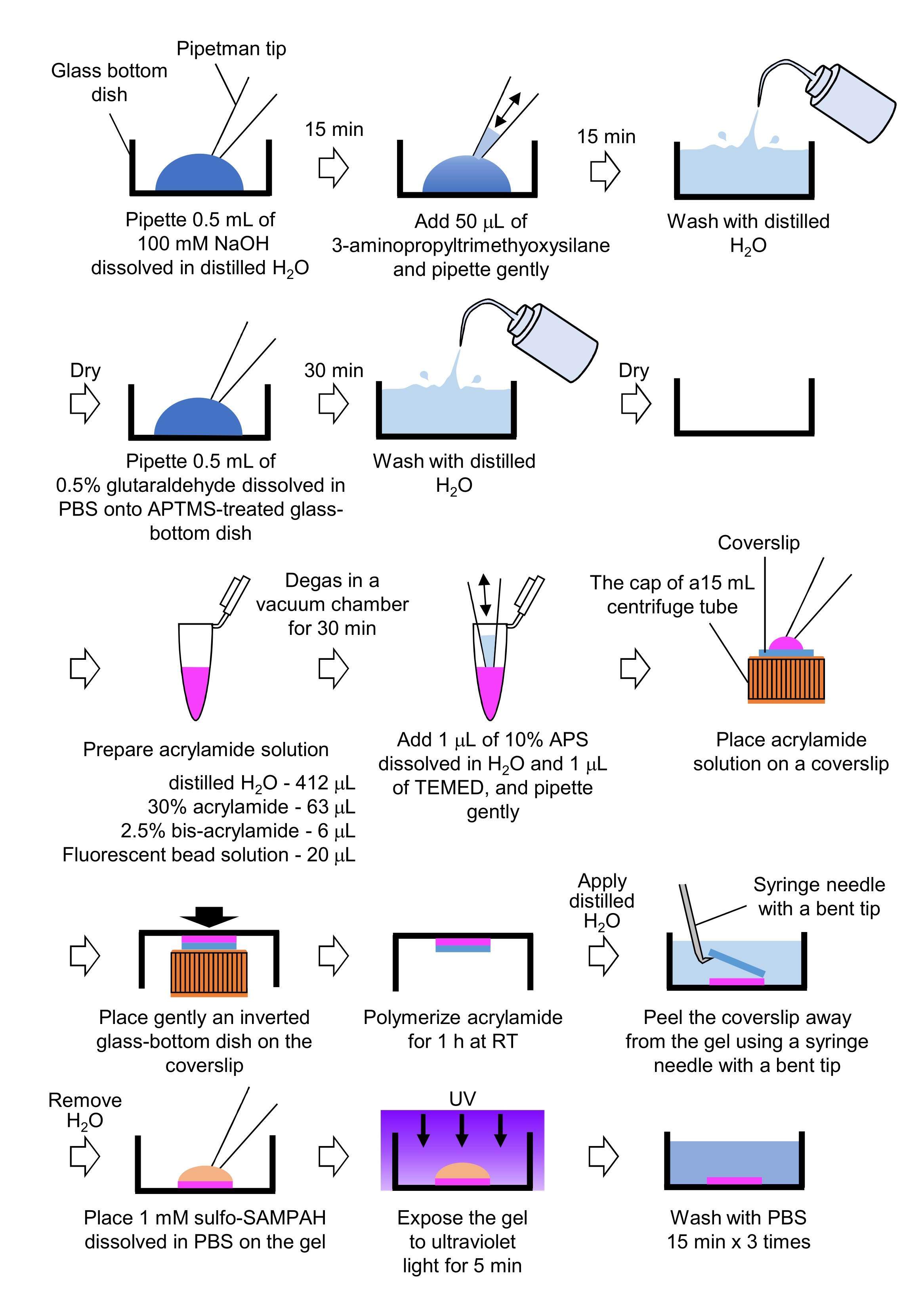
- Preparation of PAA gels required for traction force microscopy (Figure 4)
- Pipette 0.5 mL of NaOH (100 mM), dissolved in distilled H2O, on 27 mm-diameter glass-bottom dishes, and incubate for 15 min at room temperature (RT).
- Add 50 µL of 3-aminopropyltrimethyoxysilane (APTMS) into NaOH solution on the dishes and mix the solutions by gentle pipetting, and incubate for 15 min at RT.
- Wash the glass-bottom dishes with distilled H2O, and dry.
- Pipette 0.5 mL of 0.5% glutaraldehyde solution, dissolved in PBS, on the APTMS-treated glass-bottom dish, and incubate for 30 min at RT.
- Wash the glass-bottom dishes with distilled H2O and dry.
- Prepare a solution containing distilled H2O (412 µL), 30% (w/v) acrylamide solution (63 µL), 2.5% (w/v) bis-acrylamide solution (6 µL) and carboxylate-modified microsphere (fluorescent bead) solution (20 µL).
NOTE: Vortex the fluorescent bead solution to dissociate aggregated beads before adding to the acrylamide solution. - Degas the solution in a vacuum chamber for 30 min at RT.
- To this solution, add 1 µL of 10% (w/v) ammonium persulphate (APS), dissolved in distilled H2O, and 1 µL of N,N,N',N'-tetramethylethylenediamine (TEMED), and mix by gentle pipetting.
- Place a glass coverslip (18 mm diameter) on the cap of a 15 mL centrifuge tube.
- Immediately add 25 µL of the solution to the coverslip, and then gently place an inverted glass-bottom dish on the coverslip. Allow the acrylamide to polymerize for 1 h at RT.
- Apply distilled H2O to the coverslip and peel it away from the gel using a syringe needle with a bent tip.
- Remove the H2O and add sulfo-SANPAH (1 mM), dissolved in PBS, to the gel.
- Under sterile conditions, expose the gels to ultraviolet light for 5 min. Wash the gel three times with PBS for 15 min per wash.
- Determination of gel rigidity with a microsphere indentation method (Figure 5A)44.
- Transfer a microsphere which is specified for the diameter and density, to the gel and then place it on the specimen stage of a laser scanning confocal microscope.
- Focus on the gel surface and record the z-position (Figure 5B).
- Similarly, focus on the bottom of the microsphere, and record the z-position (Figure 5C).
- Calculate the indentation depth as determined by the difference between the z-positions of the gel surface and the bottom of the microsphere.
- Calculate Young's modulus E of the gel by:

where f is the buoyancy-corrected weight of the microsphere, d is the indentation depth of the gel, r is the radius of the microsphere, and v is the Poisson's ratio (whose value is 0.3, as determined previously46).
- Coat the gels with PDL and laminin as described in section 2.
- As described in sections 3 and 4, dissect out E16 mouse embryos, dissociate hippocampi, and transfect with 5 µg of pEGFP-C1 per 1.0 x 106 cells. Seed 2.0 x 105 cells on PDL/laminin-coated gels.
NOTE: For traction force analysis, microscopic imaging of fluorescently-labeled cells is necessary for determining the growth cone area.
8. Traction force microscopy at neuronal growth cones
- On DIV3, replace the culture medium with 0.5 mL of warmed Leibovitz's L-15 medium containing 2% B-27 supplement, glutamine (1 mM), penicillin (100 U/mL), and streptomycin (100 µg/mL). Maintain the neurons for 1 h at 37 °C.
- Turn on a laser scanning confocal microscope and set the stage-top incubator to 37 °C.
NOTE: The present study uses a laser scanning confocal microscope equipped with a 63x/1.2 NA water immersion objective lens and an image acquisition and analysis software (see Table of Materials). - Place the neurons in the glass-bottom dish on the pre-warmed stage-top incubator.
- Set the image acquisition parameters as follows: scan size, 512 × 512 pixels; scan area, 1.5-3x zoom; scan speed, ~1 s per frame; laser wavelength, 561 nm (the excitation wavelength for the fluorescent beads) and 488 nm (the excitation wavelength for enhanced green fluorescent protein (EGFP)); time interval, 3 s; duration, 50 frames.
- To visualize the growth cone morphology, select a growth cone that strongly expresses EGFP.
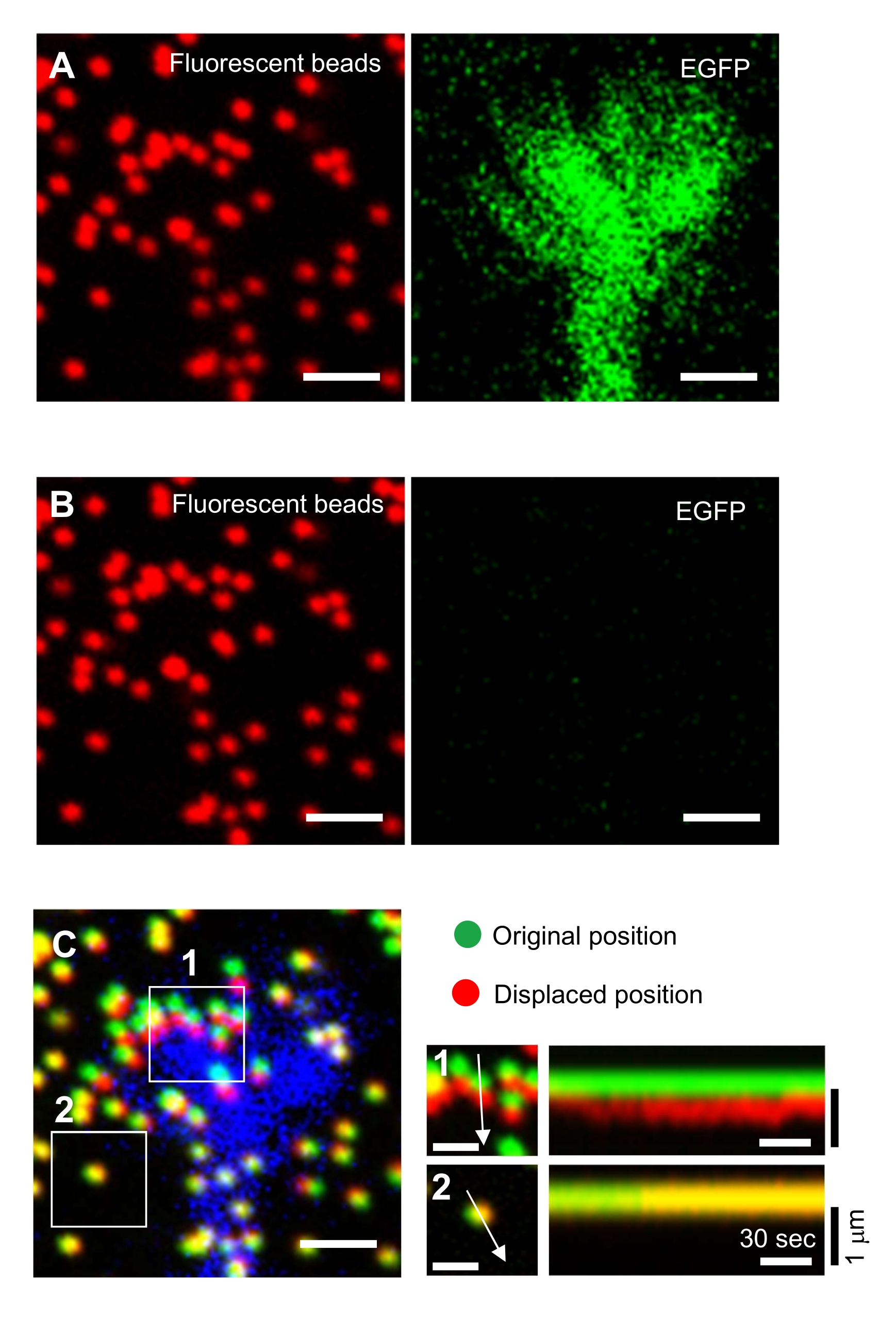
- Focus on the gel surface and acquire time-lapse images (Figure 6A and Supplemental File 4).
NOTE: For traction analysis, use two fluorescent channels (for fluorescent beads and EGFP) and a bright-field channel (capturing differential interference contrast (DIC) or phase-contrast images is recommended). - Use an image processing software (e.g., Fiji) to produce single-channel time-lapse RGB image stacks and save these images as tiff files.
NOTE: It is critical that investigators also save the raw data. - Apply 100 µL of 10% (w/v) sodium dodecyl sulfate (SDS), dissolved in distilled H2O, to the glass-bottom dish to relax the gel substrate by releasing neurons from the substrate. Incubate the dish for 5 min at 37°C to stabilize the temperature.
NOTE: The image of the beads in an unstrained substrate is used to reference the original bead positions during traction force analysis (Figure 6C). Application of SDS solution alters the xyz-positions due to thermal change in the incubator and relaxation of the cell-induced deformation. Investigators, therefore, need to correct the focal plane and x-y position. - Focus on the gel surface, and acquire an image of the beads in the unstrained substrate (Figure 6B).
- Produce a single-channel RGB image of the beads in the unstrained substrate, and save this image as a tiff file.
- Correction of the x-y position of the bead image in the unstrained substrate using Fiji
- In Fiji, open the bead image in the unstrained substrate and the time-lapse stack image of fluorescent beads.
- Select Image > Stacks > Tools > Concatenate. A dialogue box will appear on the screen (Figure 7A, 1).
- Select the bead image in the unstrained substrate and the time-lapse stack image of fluorescent beads in Image1 and Image2, respectively (Figure 7A, 2).
- Click on OK. The bead image in the unstrained substrate is added to the time-lapse stack image (Figure 7A, 3).
- Using the scroll bar (Figure 7B, 1, red frame), display the second frame of the stack image (red arrow).
- Select Plugins > StackReg (Figure 7B, 2). A dialogue box will appear on the screen (Figure 7B, 3).
- Select Rigid Body from the drop-down list (Figure 7B, 3, red frame) and click on OK. The correction of the x-y position then begins.
- Use the scroll bar to display the first frame of the x-y position-corrected stack image.
- Select Image > Duplicate (Figure 7C, 1). A dialogue box will appear on the screen (Figure 7B, 2).
- Input 1 in the Range, deselect Duplicate Stack (Figure 7C, 2), and click OK. The x-y position-corrected bead image in the unstrained substrate will appear on the screen (Figure 7C, 3). Save this image as a tiff file.
- Quantification of traction force
NOTE: The method described here for traction force analysis uses MATLAB and two MATLAB toolboxes, 'Image Processing Toolbox' and 'Parallel Computing Toolbox'. Investigators will need to install them prior to the analysis. The traction force analysis code was developed based on MATLAB version 2018a. Therefore, MATLAB version 2018a (or later) must be used for the analysis. The algorithms used for traction force analysis were described previously22.- Download the traction force analysis code TFM2021 from Supplemental File 5. Open TFM 2021 in MATLAB
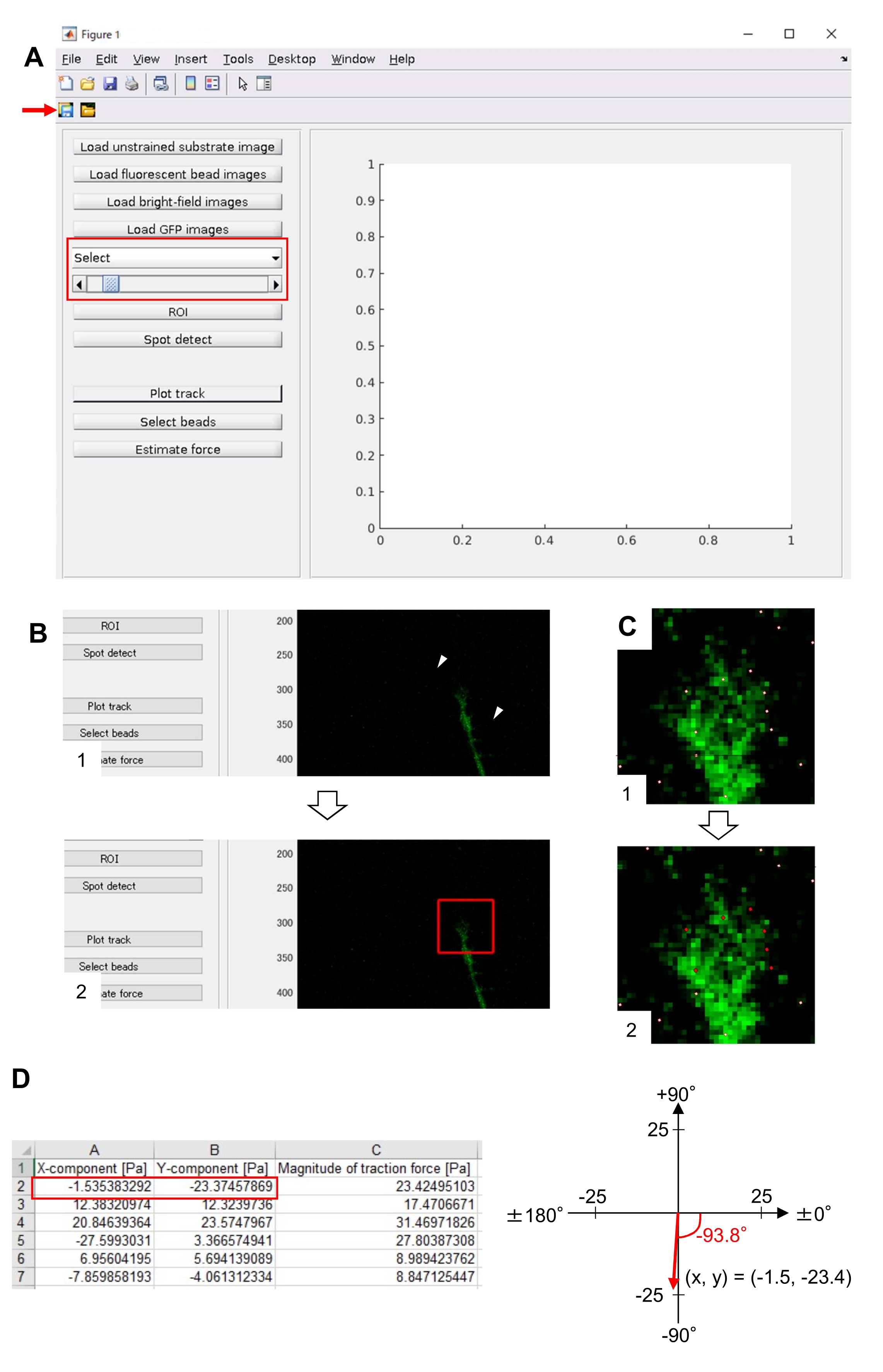
- Open main.m in TFM2021 and run it. A graphical user interface (GUI) will appear on the screen (Figure 8A).
- Click on Load unstrained substrate image and select the x-y position-corrected bead image in unstrained substrate.
- Click on Load fluorescent bead images and select the time-lapse stack image of beads.
- Click on Load bright-field images and select the time-lapse stack image of bright-field.
- Click on Load GFP Images and select the time-lapse stack image of EGFP.
- Select GFP from the drop-down list on the GUI (Figure 8A, red box).
- Click on ROI to specify the rectangle region of interest (ROI), including the growth cone, by clicking two points on the cell image displayed on the GUI (Figure 8B).
- Click on the Save button on the GUI (Figure 8A, red arrow). The selected stack images, together with the ROI, will be saved in a .mat file (MATLAB format file).
- Click on Spot detect. A dialogue box will appear on the screen.
- Input a value (usually 50-150) on the dialogue box to determine a threshold for bead detection. Clicking OK starts the calculation.
- After finishing the calculation, click on Plot track to enlarge the region selected at step 8.12.8 and display the detected beads as white dots (Figure 8C, 1).
NOTE: Confirm whether bead detection (white dots) overlaps with the fluorescent beads. Bead detection may also detect background noise. To reduce this external interference, change the threshold value at Spot detect. In addition, manually select the correct dots at step 8.12.13. - Click on Select beads, and demarcate a polygonal region that includes the correct dots under the growth cone. Press Enter on the keyboard. The white dots within the polygonal region will change into a red color (Figure 8C, 2).
- Click on Estimate force on the GUI. Then, input values for the following parameters: pixel size, µm/pixel; Young's modulus, the value calculated at Step 7.2.5; Poisson's ratio, 0.3.
- Execute Estimate force to initiate the calculation. The software will save the calculation results in a spreadsheet format file automatically.
NOTE: The spreadsheet shows the x-component, y-component, and force magnitude at every time-frame (Figure 8D). Refer to Supplemental File 6 for practice data for quantifying traction force.
Results
Single speckle imaging to quantify actin polymerization rate and clutch coupling efficiency
A high Lifeact expression permits F-actin visualization within the growth cone; a low HaloTag-actin expression allows the monitoring of F-actin retrograde flow (Figure 3, Supplemental File 2). Tracing of the actin speckles allows measurement of F-actin flow velocity (Figure 3C,D). Since the mechanical coupling of F-actin retrograde flow and the adhesive substrate reduces the F-actin flow velocity, the clutch coupling efficiency can be estimated from the velocity. Furthermore, F-actin labeling with Lifeact aids visualization of F-actin extension and is useful for quantifying the actin polymerization rate (Figure 3E,F).
Quantitative analysis of traction force
Strict adherence to the methodology presented here will reveal the movements of fluorescent beads under the growth cone (Figure 6C, Supplemental File 4). F-actin retrograde flow onto the substrate generates traction force causing the fluorescent beads to move rearward under the growth cone. The traction force analysis code estimates traction force from fluorescent bead displacement, and expresses the calculated traction force as a force vector. The direction and magnitude of the traction force are determined from the x- and y-components of the force vector (Figure 8C,D). The red box in Figure 8D represents the x- and y-components of a force vector; Figure 8C depicts the corresponding growth cone. In terms of x-y coordinates, the vector points to -93.8° against the x-axis; this orientation is directed toward the rear of the growth cone (Figure 8C). The magnitude of the traction force F was calculated as follows:


Figure 1: A growth cone machinery for force generation and growth cone navigation. A netrin-1 chemoattractant gradient induces asymmetrical stimulation of its receptor DCC on an axonal growth cone. This activates Rac1 and Cdc42, and their downstream kinase Pak1. Rac1 and Cdc42 promote (1) actin polymerization, whereas Pak1 phosphorylates shootin1, enhancing shootin1-mediated (2) clutch coupling. The asymmetric activation of actin dynamics and the clutch coupling within the growth cone increases (3) traction force on the side of the netrin-1 source, thereby generating a directional driving force for growth cone attraction. The protocols presented here allow for the quantification of the key variables (1)-(3) for growth cone navigation. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Fluorescence images of a neuronal growth cone under a fully opened and narrowed diaphragm. The growth cone expresses Lifeact and HaloTag-actin. (A) A high Lifeact expression permits visualization of growth cone morphology. On the other hand, HaloTag-actin expression levels are very low, with dim signals when the diaphragm is fully opened. (B) When the diaphragm is appropriately narrowed, background signals diminish, and single actin speckles appear in the growth cone. Scale bars: 5 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Steps for quantifying F-actin flow velocity and actin polymerization rate using an image processing and analysis software Fiji. (A) Adjust the angle of the image for analysis. (1) Find and select an actin speckle (arrowhead) that flows in an F-actin bundle. (2) Select Image > Transform > Rotate. (3) Set the angle so that the F-actin bundle is directed upward. (4) The angle of the image will be changed. (B) Demarcate the region, including the actin speckle and the F-actin bundle. (1) Click on Rectangle on the toolbar. (2) Delineate a region on the image. The brightness and contrast of the image are increased to allow clear visualization of the actin speckle and the tip of the F-actin bundle. (3) Select Image > Duplicate. (4) Input the five time-frames that show actin speckle flow in the F-actin bundle. (5) The selected stack image will appear on the screen. (C) Overlay circles on the actin speckle. (1) Click on Oval on the toolbar. (2) Draw a circle on an actin speckle. (3) Select Image > Overlay > Add Selection. (4) The circle is overlaid. 5) Repeat overlaying the circles on the remaining actin speckles. (D) Measure the translocation distance of actin speckles during the five time-frames. (1) Click on Straight on the toolbar. (2) Draw a line which links the centers of the circles. (3) Select Analyze > Measure. The result, indicated by the parameter Height (red box), relaying the actin speckle translocation distance. (E) Measure the change in the length of F-actin protrusion during the five time-frames. (1) Draw a line which links the tips of the F-actin protrusion. (2) Select Analyze > Measure. The result (red box), Height, indicates the extension length of the F-actin bundle. (F) The actin polymerization rate is calculated from the sum of the F-actin flow velocity and the extension rate. See also Supplemental File 2. Supplemental File 3 assists investigators to practice the methodology described above. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Steps for the PAA gel preparation. Please see step 7.1 for a detailed description. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Determination of PAA gel rigidity. (A) A microsphere indentation method. When a microsphere is placed on a fluorescent bead-embedded PAA gel, the weight of the microsphere causes an indentation in the gel. The indentation depth is calculated by subtracting the z-positions of the PAA gel surface from the bottom of the microsphere (B,C) Fluorescence images of a PAA gel indented by a microsphere and containing fluorescent beads. A laser scanning confocal microscope was used to capture images of the gel surface (B) and the bottom of the microsphere (C). Signals from the fluorescent beads are not visible at the gel surface in the indented region (B, circle). They can, however, be observed at the bottom of the microsphere. Scale bars: 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Force mapping of a neuronal growth cone. (A-C) Fluorescence images of beads embedded in a PAA gel and a neural growth cone visualized with EGFP. Following the acquisition of time-lapse images (A), the neuron was released from the gel substrate by applying SDS solution, and an image of the beads in the unstrained substrate was captured (B). (C) The image of the beads in the unstrained substrate shows the beads in their original (green) and displaced (red) positions. The EGFP signal of the growth cone is shown in blue. Kymographs (right) show the movements of beads at 3 s intervals for a duration of 147 s, indicated by the arrows in boxed areas 1 and 2. The bead in area 2 is a reference bead. Scale bars: 2 µm for (A), (B) and (C, left), and 1 µm for (C, middle). See also Supplemental File 4. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Steps for the correcting x-y position of the bead image in unstrained substrate using Fiji. (A) Concatenate the bead image in unstrained substrate with the time-lapse stack image of fluorescent beads. (1) Select Image > Stacks > Tools > Concatenate. (2) Select the beads image in unstrained substrate and the time-lapse stack image of fluorescent beads in Image1 and Image2, respectively. Click on OK. (3) The bead image in the unstrained substrate is added to the time-lapse stack image. (B) Correct the x-y position of the fluorescent bead image. (1) Use the scroll bar (red frame) to select the second frame (red arrow) in the image stack. (2) Select Plugins > StackReg. (3) Select Rigid Body from the drop-down list (red frame) and click on OK. The correction of the x-y position will begin. (C) Save the x-y position-corrected bead image. After selecting the first frame of the x-y position-corrected stack image, (1) select Image > Duplicate. (2) input 1 into the Range and deselect Duplicate stack. Then click on OK. (3) the x-y position-corrected bead image in the unstrained substrate will appear on the screen. Save this image as a tiff file. Supplemental File 6 assists investigators to practice the methodology described above. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: Analysis of traction force under a neuronal growth cone using an open-source traction force analysis code. (A) GUI for analyzing traction force. Time-lapse images selected on the GUI can be confirmed from the drop-down list and with the slider indicated (red box). (B) Select a region that includes the growth cone. (1) Click on ROI on the GUI. With the mouse cursor, specify two points (arrowheads) on the cell image. (2) A red box will appear on the cell image. Two clicks determine the locations of two corners. (C) Select the detected beads (white dots) under the growth cone. (1) On the GUI, click on Select beads, and demarcate a polygonal region that includes the growth cone by clicking. Press the Enter key. (2) The white dots within the polygonal region will change into a red color. (D) Calculated results of the direction and magnitude of the traction force. The red box in the spreadsheet represents the x- and y-components of the force vector, estimated by traction force analysis. On the x-y coordinate in the right panel, the force vector generated by the growth cone points to -93.8° against the x-axis; this orientation is directed toward the rear of the growth cone in (C). Supplemental File 6 assists investigators to practice the methodology described above. Please click here to view a larger version of this figure.
{kind=link}
Supplemental File 1: Recipes of solutions and media used in this study. See text for detailed usage. Please click here to download this File.
Supplemental File 2: Fluorescence imaging of Lifeact and fluorescent speckle imaging of HaloTag-actin in a nerve growth cone. Lifeact (green) and HaloTag-actin (magenta). Images were acquired every 3 s for a total duration of 147 s. Scale bar: 2 µm. See also Figure 3. Please click here to download this File.
Supplemental File 3: Practice data for quantifying F-actin flow velocity and the actin polymerization rate. A multichannel time-lapse stack image of Lifeact (green) and HaloTag-actin (magenta). See also Figure 3. Please click here to download this File.
Supplemental File 4: A force-mapping video detecting traction force at a neuronal growth cone. The original (green) and displaced (red) positions of the beads. The EGFP signal in the growth cone is shown in blue. Images were acquired every 3 s for 147 s. Scale bar: 2 µm. See also Figure 6. Please click here to download this File.
Supplemental File 5: Traction force analysis code. Please see Step 8.12 for detailed usage. Please click here to download this File.
Supplemental File 6: Practice data for quantifying traction force. A single-channel RGB image of the beads in unstrained substrate and single-channel time-lapse stack RGB images of fluorescent beads under a growth cone, EGFP, and bright-field. See also Figures 7 and 8. Please click here to download this File.
Discussion
The protocols described in this study use commercially available materials and microscopy equipment routinely found in all the laboratories, institutes, and universities. Therefore, investigators can easily adopt the present single speckle imaging and traction force microscopy in their studies.
The speckle imaging can analyze actin polymerization and clutch coupling. In addition, speckle imaging can monitor the retrograde flow of clutch molecules such as shootin1 and cortactin, which interact with F-actin retrograde flow. By using a TIRF microscope, the retrograde flow of the cell adhesion molecule L1-CAM can also be monitored23,41; L1-CAM undergoes grip and slip behaviors that reflect the clutch coupling efficiency23,41. Although the present study employs the TMR-HaloTag system for speckle imaging, other fluorescent proteins, such as EGFP and monomeric red fluorescent protein, are also available in the analysis16,18,20,23,24,27,39. The essentials for visualizing actin speckles are a low expression level of fluorescent actin and the illumination of a minimum area (Figure 2). In this protocol, Lifeact and HaloTag-actin signals are sequentially acquired. Because actin retrograde flow is relatively slow (4.5 ± 0.1 µm/min)24, analysis of F-actin retrograde flow and actin polymerization are unaffected by sequential image acquisition of different fluorescent channels (~1 s interval). Lifeact is a widely-used F-actin marker, but can compete with actin binding proteins47. Of further importance, Lifeact can alter actin dynamics, thereby affecting F-actin structures and the cell morphology47,48,49.
Traction force microscopy can detect forces to drive growth cone advance. By mounting the neurons within an extracellular matrix, investigators can also analyze forces generated in a semi-3D environment11. High-magnification imaging is important for accurate quantification of traction force because growth cones generate weak traction forces7. Although other methods with nanopillars or stress-sensitive biosensors are also used to measure traction force50,51, PAA gel-based method is highly adaptable and allows for the adjustment of substrate rigidity by varying the concentrations of acrylamide and bis-acrylamide41,44,52. In this protocol, the PAA gel is prepared at a final concentration of 3.75% acrylamide and 0.03% bis-acrylamide; Young's modulus is ~270 Pa22 and this stiffness is within the range of brain tissue (100-10,000 Pa)53,54,55. Due to the thickness of the PAA gel (~100 µm), this method limits the use of high-magnification lenses during microscopy. To obtain high-magnification images, investigators should use the zoom function in a laser scanning confocal microscope.
In conclusion, the present speckle imaging and traction force microscopy enable quantitative analyses of the key events in force generations. This information will be invaluable for improving understanding of the mechanisms that underlie growth cone advancement and navigation.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This research was supported in part by AMED under grant number 21gm0810011h0005 (N.I. and Y.S.), JSPS KAKENHI (JP19H03223, N.I.) and JSPS Grants-in-Aid for Early-Career Scientists (JP19K16258, T.M.), the Osaka Medical Research Foundation for Incurable Diseases (T.M.), and NAIST Next Generation Interdisciplinary Research Project (Y.S.).
Materials
| Name | Company | Catalog Number | Comments |
| 0.5% trypan blue stain solution | Nacalai | 29853-34 | |
| 3-aminopropyltrimethyoxysilane | Sigma | 281778-100ML | |
| Acrylamide monomer | Nacalai | 00809-85 | |
| Ammonium persulphate | Cytiva | 17-1311-1 | |
| Axio Observer Z1 | Zeiss | 431007-9902-000 | Epi-fluorescence microscope (single speckle imaging) |
| B-27 supplement (50x) | Thermo Fisher Scientific | 17504-044 | |
| Bovine serum albmine | Sigma | A7906-10G | |
| C-Apochromat 63x/1.2 W Corr | Zeiss | 421787-9970-799 | Objective lens (traction force microscopy) |
| Coverslip (diameter 18 mm) | Matsunami | C018001 | |
| D-glucose | Nacalai | 16806-25 | |
| DNaseI | Sigma | DN25-100MG | |
| Fetal bovine serum | Thermo Fisher Scientific | 10270-106 | |
| Fiji | Open source software package | https://imagej.net/software/fiji/ | |
| FluoSpheres carboxylate-modified 0.2 mm, red (580/605), 2% solid | Thermo Fisher Scientific | F8810 | carboxylate-modified microspheres |
| Glass bottom dish (14 mm diameter) | Matsunami | D1130H | |
| Glass bottom dish (27 mm diameter) | Matsunami | D1140H | |
| Glutaraldehyde solution | Sigma | G5882-10X10ML | |
| HaloTag TMR ligand | Promega | G8251 | |
| HBO103 W/2 | Osram | 4050300382128 | Mercury lamp (single speckle imaging) |
| Image Processing Toolbox | MathWork | https://www.mathworks.com/products/image.html | |
| Laminin solution from mouse EHS tumor | Wako | 120-05751 | |
| Leibovitz’s L-15 medium | Thermo Fisher Scientific | 11415064 | |
| L-glutamine | Nacalai | 16919-42 | |
| LSM710 | Zeiss | N/A | Conforcal laser microscope (traction force microscopy) |
| MATLAB2018a | MathWork | https://www.mathworks.com/products/new_products/release2018a.html | |
| Mouse C57BL/6 | Japan SLC | N/A | |
| Mouse neuron nucleofector kit | Lonza | VPG-1001 | |
| N,N,N’,N’-tetramethylethylenediamine (TEMED) | Nacalai | 33401-72 | |
| N,N’-methylenebisacrylamide | Nacalai | 22402-02 | |
| Neurobasal medium | Thermo Fisher Scientific | 21103-049 | |
| Nucleofector I | Amaxa | AAD-1001 | Electroporation apparatus |
| ORCA Flash 4.0 V2 | Hamamatsu | C11440-22CU | CMOS camera (single speckle imaging) |
| Papain | Nacalai | 26036-34 | |
| Parallel Computing Toolbox | MathWork | https://www.mathworks.com/products/parallel-computing.html | |
| pEGFP-C1 | Clontech | 1528177 | |
| Penicillin-streptomycin (100x) | Nacalai Tesque | 26253-84 | |
| pFN21A-HaloTag-actin | (Minegishi et al., 2018) | N/A | |
| Phosphate buffered saline (PBS) pH 7.4 (10x) | Thermo Fisher Scientific | 70011-044 | |
| Plan-Apochromat 100x/1.4 Oil | Zeiss | 420790-9901-000 | Objective lens (single speckle imaging) |
| pmNeonGreen-N1-Lifeact | (Kastian et al., 2021) | N/A | |
| Poly-D-lysine hydrobromide | Sigma | P6407-5MG | |
| Slulfo-SAMPHA | Thermo Fisher Scientific | 22589 | |
| Sodium dodecyl sulfate | Nacalai | 08933-05 | |
| Sodium hydrate (NaOH) | Nacalai | 31511-05 | |
| Steel ball | Sako tekkou | N/A | Microshpere to determin PAA gel rigidity. 0.6 mm diameter, 7.87 g/cm3. |
| ZEN2009 | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html#introduction | Image acquisition software (traction force microscopy) |
| ZEN2012 | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html#introduction | Image acquisition software (single speckle imaging) |
References
- TessierLavigne, M., Goodman, C. S. The molecular biology of axon guidance. Science. 274, 1123-1133 (1996).
- Marin, O., Valiente, M., Ge, X., Tsai, L. H. Guiding neuronal cell migrations. Cold Spring Harbor Perspectives in Biology. 2, 001834 (2010).
- Vitriol, E. A., Zheng, J. Q. Growth cone travel in space and time: the cellular ensemble of cytoskeleton, adhesion, and membrane. Neuron. 73 (6), 1068-1081 (2012).
- Cooper, J. A. Cell biology in neuroscience: mechanisms of cell migration in the nervous system. Journal of Cell Biology. 202, 725-734 (2013).
- Dupraz, S., et al. RhoA controls axon extension independent of specification in the developing brain. Current Biology. 29, 3874-3886 (2019).
- Suter, D. M., Miller, K. E. The emerging role of forces in axonal elongation. Progress in Neurobiology. 94, 91-101 (2011).
- Franze, K. Integrating chemistry and mechanics: the forces driving axon growth. Annual Review of Cell and Developmental Biology. 36, 61-83 (2020).
- Mitchison, T., Kirschner, M. Cytoskeletal dynamics and nerve growth. Neuron. 1, 761-772 (1988).
- Suter, D. M., Forscher, P. Substrate-cytoskeletal coupling as a mechanism for the regulation of growth cone motility and guidance. Journal of Neurobiology. 44, 97-113 (2000).
- Lowery, L. A., Van Vactor, D. The trip of the tip: understanding the growth cone machinery. Nature Reviews Molecular Cell Biology. 10, 332-343 (2009).
- Minegishi, T., et al. Shootin1b mediates a mechanical clutch to produce force for neuronal migration. Cell Reports. 25, 624-639 (2018).
- Minegishi, T., Inagaki, N. Forces to drive neuronal migration steps. Frontiers in Cell and Developmental Biology. 8, 863 (2020).
- Forscher, P., Smith, S. J. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. Journal of Cell Biology. 107, 1505-1516 (1988).
- Suter, D. M., Forscher, P. An emerging link between cytoskeletal dynamics and cell adhesion molecules in growth cone guidance. Current Opinion in Neurobiology. 8, 106-116 (1998).
- Pollard, T. D., Borisy, G. G. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112, 453-465 (2003).
- Waterman-Storer, C. M., Desai, A., Bulinski, J. C., Salmon, E. D. Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Current Biology. 8, 1227-1230 (1998).
- Katoh, K., Hammar, K., Smith, P. J., Oldenbourg, R. Birefringence imaging directly reveals architectural dynamics of filamentous actin in living growth cones. Molecular Biology of the Cell. 10, 197-210 (1999).
- Watanabe, N., Mitchison, T. J. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science. 295, 1083-1086 (2002).
- Medeiros, N. A., Burnette, D. T., Forscher, P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nature Cell Biology. 8, 215-226 (2006).
- Shimada, T., et al. Shootin1 interacts with actin retrograde flow and L1-CAM to promote axon outgrowth. Journal of Cell Biology. 181, 817-829 (2008).
- He, M., Zhang, Z. H., Guan, C. B., Xia, D., Yuan, X. B. Leading tip drives soma translocation via forward F-actin flow during neuronal migration. Jounal of Neuroscience. 30, 10885-10898 (2010).
- Toriyama, M., Kozawa, S., Sakumura, Y., Inagaki, N. Conversion of a signal into forces for axon outgrowth through Pak1-mediated shootin1 phosphorylation. Current Biology. 23, 529-534 (2013).
- Abe, K., et al. Grip and slip of L1-CAM on adhesive substrates direct growth cone haptotaxis. Proceedings of the National Academy of Sciences of the United States of America. 115, 2764-2769 (2018).
- Baba, K., et al. Gradient-reading and mechano-effector machinery for netrin-1-induced axon guidance. Elife. 7, 34593 (2018).
- Nichol, R. I., Hagen, K. M., Lumbard, D. C., Dent, E. W., Gomez, T. M. Guidance of axons by local coupling of retrograde flow to point contact adhesions. Journal of Neuroscience. 36, 2267-2282 (2016).
- Shekarabi, M., et al. Deleted in colorectal cancer binding netrin-1 mediates cell substrate adhesion and recruits Cdc42, Rac1, Pak1, and N-WASP into an intracellular signaling complex that promotes growth cone expansion. Journal of Neuroscience. 25, 3132-3141 (2005).
- Kubo, Y., et al. Shootin1-cortactin interaction mediates signal-force transduction for axon outgrowth. Journal of Cell Biology. 210, 663-676 (2015).
- Huber, A. B., Kolodkin, A. L., Ginty, D. D., Cloutier, J. F. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annual Review of Neuroscience. 26, 509-563 (2003).
- Kolodkin, A. L., Tessier-Lavigne, M. Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harbor Perspectives in Biology. 3, 001727 (2011).
- Ayala, R., Shu, T., Tsai, L. H. Trekking across the brain: the journey of neuronal migration. Cell. 128, 29-43 (2007).
- Tatavarty, V., Kim, E. J., Rodionov, V., Yu, J. Investigating sub-spine actin dynamics in rat hippocampal neurons with super-resolution optical imaging. PLoS One. 4, 7724 (2009).
- Frost, N. A., Shroff, H., Kong, H., Betzig, E., Blanpied, T. A. Single-molecule discrimination of discrete perisynaptic and distributed sites of actin filament assembly within dendritic spines. Neuron. 67, 86-99 (2010).
- Chazeau, A., et al. Nanoscale segregation of actin nucleation and elongation factors determines dendritic spine protrusion. EMBO Journal. 33, 2745-2764 (2014).
- Garcia, M., et al. Two-tiered coupling between flowing actin and immobilized N-cadherin/catenin complexes in neuronal growth cones. Proceedings of the National Academy of Sciences of the United States of America. 112, 6997-7002 (2015).
- Swaminathan, V., et al. Actin retrograde flow actively aligns and orients ligand-engaged integrins in focal adhesions. Proceedings of the National Academy of Sciences of the United States of America. 114, 10648-10653 (2017).
- Tsai, T. Y., et al. Efficient front-rear coupling in neutrophil chemotaxis by dynamic myosin II localization. Developmental Cell. 49, 189-205 (2019).
- Zhang, X. F., et al. Regulation of axon growth by myosin II-dependent mechanocatalysis of cofilin activity. Journal of Cell Biology. 218 (7), 2329-2349 (2019).
- Reversat, A., et al. Cellular locomotion using environmental topography. Nature. 582, 582-585 (2020).
- Katsuno, H., et al. Actin migration driven by directional assembly and disassembly of membrane-anchored actin filaments. Cell Reports. 12, 648-660 (2015).
- Urasaki, A., et al. Shootins mediate collective cell migration and organogenesis of the zebrafish posterior lateral line system. Scientific Reports. 9, 12156 (2019).
- Abe, K., et al. Mechanosensitive axon outgrowth mediated by L1-laminin clutch interface. Biophysical Journal. 120, 3566-3576 (2021).
- Kastian, R. F., et al. Shootin1a-mediated actin-adhesion coupling generates force to trigger structural plasticity of dendritic spines. Cell Reports. 35, 109130 (2021).
- Riedl, J., et al. Lifeact: a versatile marker to visualize F-actin. Nature Methods. 5, 605-607 (2008).
- Chan, C. E., Odde, D. J. Traction dynamics of filopodia on compliant substrates. Science. 322, 1687-1691 (2008).
- Wang, Y. L., Pelham, R. J. Preparation of a flexible, porous polyacrylamide substrate for mechanical studies of cultured cells. Methods in Enzymology. 298, 489-496 (1998).
- Li, Y., Hu, Z., Li, C. New method for measuring poisson's ratio in polymer gels. Journal of Applied Polymer Science. 50, 1107-1111 (1993).
- Belyy, A., Merino, F., Sitsel, O., Raunser, S. Structure of the Lifeact-F-actin complex. PLoS Biology. 18, 3000925 (2020).
- Flores, L. R., Keeling, M. C., Zhang, X., Sliogeryte, K., Gavara, N. Lifeact-GFP alters F-actin organization, cellular morphology and biophysical behaviour. Scientific Reports. 9, 3241 (2019).
- Kumari, A., Kesarwani, S., Javoor, M. G., Vinothkumar, K. R., Sirajuddin, M. Structural insights into actin filament recognition by commonly used cellular actin markers. EMBO Journal. 39, 104006 (2020).
- du Roure, O., et al. Force mapping in epithelial cell migration. Proceedings of the National Academy of Sciences of the United States of America. 102, 2390-2395 (2005).
- Grashoff, C., et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 466, 263-266 (2010).
- Koch, D., Rosoff, W. J., Jiang, J., Geller, H. M., Urbach, J. S. Strength in the periphery: growth cone biomechanics and substrate rigidity response in peripheral and central nervous system neurons. Biophysical Journal. 102 (3), 452-460 (2012).
- Barnes, J. M., Przybyla, L., Weaver, V. M. Tissue mechanics regulate brain development, homeostasis and disease. Journal of Cell Science. 130, 71-82 (2017).
- Moore, S. W., Roca-Cusachs, P., Sheetz, M. P. Stretchy proteins on stretchy substrates: the important elements of integrin-mediated rigidity sensing. Developmental Cell. 19, 194-206 (2010).
- Tyler, W. J. The mechanobiology of brain function. Nature Reviews Neuroscience. 13, 867-878 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved