Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Stratégies d’inoculation pour infecter les racines des plantes avec des micro-organismes transmis par le sol
Dans cet article
Résumé
Ce protocole présente un résumé détaillé des stratégies pour inoculer les racines des plantes avec des microbes transmis par le sol. Exemple pour les champignons Verticillium longisporum et Verticillium dahliae, trois systèmes d’infection racinaire différents sont décrits. Les applications potentielles et les analyses en aval possibles sont mises en évidence, et les avantages ou les inconvénients sont discutés pour chaque système.
Résumé
La rhizosphère abrite une communauté microbienne très complexe dans laquelle les racines des plantes sont constamment remises en question. Les racines sont en contact étroit avec une grande variété de micro-organismes, mais les études sur les interactions dans le sol sont encore en retard sur celles effectuées sur les organes en surface. Bien que certaines stratégies d’inoculation pour infecter les plantes modèles avec des agents pathogènes racinaires modèles soient décrites dans la littérature, il reste difficile d’obtenir un aperçu méthodologique complet. Pour résoudre ce problème, trois systèmes d’inoculation racinaire différents sont décrits avec précision qui peuvent être appliqués pour mieux comprendre la biologie des interactions racine-microbe. À titre d’illustration, les espèces de Verticillium (à savoir , V. longisporum et V. dahliae) ont été utilisées comme agents pathogènes modèles envahissant les racines. Cependant, les méthodes peuvent être facilement adaptées à d’autres microbes colonisateurs de racines - à la fois pathogènes et bénéfiques. En colonisant le xylème végétal, les champignons vasculaires transmis par le sol tels que Verticillium spp. présentent un mode de vie unique. Après l’invasion racinaire, ils se propagent par les vaisseaux du xylème par voie acropétale, atteignent la pousse et provoquent des symptômes de la maladie. Trois espèces végétales représentatives ont été choisies comme hôtes modèles : Arabidopsis thaliana, colza économiquement important (Brassica napus) et tomate (Solanum lycopersicum). Des protocoles étape par étape sont donnés. Des résultats représentatifs des tests de pathogénicité, des analyses transcriptionnelles des gènes marqueurs et des confirmations indépendantes par des constructions de rapporteurs sont présentés. En outre, les avantages et les inconvénients de chaque système d’inoculation sont discutés en profondeur. Ces protocoles éprouvés peuvent aider à fournir des approches pour les questions de recherche sur les interactions racine-microbe. Savoir comment les plantes font face aux microbes dans le sol est crucial pour développer de nouvelles stratégies visant à améliorer l’agriculture.
Introduction
Les sols naturels sont habités par un nombre étonnant de microbes qui peuvent être neutres, nocifs ou bénéfiques pour les plantes1. De nombreux agents pathogènes des plantes sont transmis par le sol, entourent les racines et attaquent l’organe souterrain. Ces micro-organismes appartiennent à une grande variété de clades : champignons, oomycètes, bactéries, nématodes, insectes et certains virus 1,2. Une fois que les conditions environnementales favorisent l’infection, les plantes sensibles deviennent malades et les rendements des cultures diminuent. Les effets du changement climatique, tels que le réchauffement de la planète et les conditions météorologiques extrêmes, augmenteront la proportion d’agents pathogènes des plantes transmis par le sol3. Par conséquent, il deviendra de plus en plus important d’étudier ces microbes destructeurs et leur impact sur la production de denrées alimentaires et d’aliments pour animaux, mais aussi sur les écosystèmes naturels. De plus, il y a des mutualistes microbiens dans le sol qui interagissent étroitement avec les racines et favorisent la croissance, le développement et l’immunité des plantes. Lorsqu’elles sont confrontées à des agents pathogènes, les plantes peuvent recruter activement des adversaires spécifiques dans la rhizosphère qui peuvent soutenir la survie de l’hôte en supprimant les agents pathogènes 4,5,6,7. Cependant, les détails mécanistes et les voies impliquées dans les interactions bénéfiques racine-microbe sont souvent encore inconnus6.
Il est donc essentiel d’élargir la compréhension générale des interactions racine-microbe. Des méthodes fiables d’inoculation des racines avec des micro-organismes transmis par le sol sont nécessaires pour effectuer des études de modèles et transférer les résultats à des applications agricoles. Les interactions bénéfiques dans le sol sont étudiées, par exemple, avec Serendipita indica (anciennement connu sous le nom de Piriformospora indica), Rhizobium spp. fixateur d’azote ou champignons mycorhiziens, tandis que les agents pathogènes des plantes connus dans le sol comprennent Ralstonia solanacearum, Phytophthora spp., Fusarium spp. et Verticillium spp.1. Ces deux derniers sont des genres fongiques qui sont répartis dans le monde entier et causent des maladies vasculaires2. Verticillium spp. (Ascomycota) peut infecter des centaines d’espèces végétales - principalement des dicotylédones, y compris des annuelles herbacées, des plantes vivaces ligneuses et de nombreuses plantes cultivées 2,8. Les hyphes de Verticillium pénètrent dans la racine et se développent à la fois intercellulairement et intracellulairement vers le cylindre central pour coloniser les vaisseaux du xylème 2,9. Dans ces vaisseaux, le champignon reste pendant la majeure partie de son cycle de vie. Comme la sève du xylème est pauvre en nutriments et transporte des composés de défense des plantes, le champignon doit s’adapter à cet environnement unique. Ceci est accompli par la sécrétion de protéines liées à la colonisation qui permettent à l’agent pathogène de survivre dans son hôte10,11. Après avoir atteint le système vasculaire racinaire, le champignon peut se propager dans les vaisseaux du xylème par voie acropétale jusqu’au feuillage, ce qui conduit à une colonisation systémique de l’hôte 9,12. À ce stade, la plante est affectée négativement dans la croissance 9,10,13. Par exemple, un retard de croissance et des feuilles jaunes se produisent ainsi qu’une sénescence prématurée 13,14,15,16.
Un membre de ce genre est Verticillium longisporum, qui est très adapté aux hôtes brassicacés, tels que le colza agronomiquement important, le chou-fleur et la plante modèle Arabidopsis thaliana12. Plusieurs études ont combiné V. longisporum et A. thaliana pour obtenir des connaissances approfondies sur les maladies vasculaires transmises par le sol et les réponses de défense racinaire qui en résultent 13,15,16,17. Des tests de susceptibilité simples peuvent être réalisés en utilisant le système modèle V. longisporum / A. thaliana et des ressources génétiques bien établies sont disponibles pour les deux organismes. Étroitement lié à V. longisporum est l’agent pathogène Verticillium dahliae. Bien que les deux espèces fongiques effectuent un style de vie vasculaire et un processus d’invasion similaires, leur efficacité de propagation des racines aux feuilles et les symptômes de la maladie provoquée chez A. thaliana sont différents: alors que V. longisporum induit généralement une sénescence précoce, l’infection à V. dahliae entraîne un flétrissement18. Récemment, un résumé méthodologique a présenté différentes stratégies d’inoculation racinaire pour infecter A. thaliana avec V. longisporum ou V. dahliae, aidant à planifier des configurations expérimentales19. Sur le terrain, V. longisporum cause parfois des dommages importants dans la production de colza12, tandis que V. dahliae a une très large gamme d’hôtes comprenant plusieurs espèces cultivées, telles que la vigne, la pomme de terre et la tomate8. Cela rend les deux agents pathogènes économiquement intéressants à étudier.
Ainsi, les protocoles suivants utilisent à la fois V. longisporum et V. dahliae comme agents pathogènes racines modèles pour illustrer les approches possibles pour les inoculations racinaires. Arabidopsis (Arabidopsis thaliana), colza (Brassica napus) et tomate (Solanum lycopersicum) ont été choisis comme hôtes modèles. Des descriptions détaillées des méthodologies peuvent être trouvées dans le texte ci-dessous et la vidéo qui l’accompagne. Les avantages et les inconvénients de chaque système d’inoculation sont discutés. Dans l’ensemble, cette collection de protocoles peut aider à identifier une méthode appropriée pour des questions de recherche spécifiques dans le contexte des interactions racine-microbe.
Protocole
1. Milieux pour cultures fongiques et systèmes d’inoculation végétale
- Bouillon liquide de dextrose de pomme de terre (PDB) : Préparer 21 g/L de PDB dans de l’eau ultrapure dans une fiole thermostable.
- Bouillon liquide de dextrose Czapek (CDB) : Préparer 42 g/L de CDB dans de l’eau ultrapure dans une fiole thermostable.
- Milieu pour le système d’inoculation de la boîte de Petri : Préparer une fiole thermostable avec 1,5 g/L de milieu Murashige et Skoog (MS) et 8 g/L d’agar dans de l’eau ultrapure.
REMARQUE: Évitez le sucre dans ce milieu car il entraînera une croissance fongique excessive après l’inoculation. - Milieu pour le système d’inoculation à base de gobelet en plastique : Préparer une fiole thermostable contenant 4,4 g/L de MS, 0,2 g/L mgSO4, 1 g/L deKNO3, 0,5 g/L d’acide 2-(N-morpholino)éthanesulfonique (MES) et 6,0 g/L de gélose dans de l’eau ultrapure et ajuster le pH à 5,7 avec 5 M KOH.
REMARQUE: Évitez le sucre dans ce milieu car il entraînera une croissance fongique excessive après l’inoculation. - Milieu 1/4 MS : Préparer 1,2 g/L de SEP dans de l’eau ultrapure.
- Utilisez l’autoclave pour stériliser toutes les solutions ci-dessus. Mettez les flacons en verre dans le panier, fermez le couvercle et stérilisez pendant 15 min à 121 °C et 98,9 kPa.
2. Stérilisation de la surface des graines de plantes
REMARQUE: Utilisez toujours le protocole ci-dessous pour stériliser la surface des graines d’Arabidopsis, de colza et de tomate avant le semis.
- Transférer les graines dans un tube de réaction de 2 mL. Placez le tube dans un exsiccateur d’une capacité interne de 5,8 L.
- Générer du chlore gazeux dans l’exsiccateur en ajoutant 6 mL d’acide chlorhydrique à 33 % (HCl) dans 100 mL d’hypochlorite de sodium aqueux (NaClO) à 12 %.
- Fermez immédiatement le couvercle de l’exsiccateur et incubez les graines pendant 3 h dans le gaz.
3. Préparation de l’inoculum avec des spores de Verticillium (conidies dérivées asexuées)
REMARQUE: Cultiver V. dahliae (souche JR2) de la même manière que V. longisporum (souche Vl43)17,18,19. Assurez-vous que tout l’équipement et les milieux sont exempts de germes et que toutes les étapes sont effectuées dans une hotte à flux laminaire pour garder l’inoculum axenique.
- Remplir 150 mL de PDB liquide (étape 1.1) dans une fiole de chicanerie de 500 mL et compléter le milieu avec 500 mg/L de céfotaxime.
- Ajouter Verticillium conida du stockage du stock de glycérol au milieu PDB. Fermez la fiole avec un bouchon en mousse stérile.
- Incuber la culture pendant 7 à 10 jours dans une boîte sombre à température ambiante (RT) sous agitation continue et horizontale (agitateur rotatif; 60 tr / min). Il en résulte de petites sphères de mycélium blanches.
- Retirez et jetez soigneusement le surnageant PDB. La plupart des mycéliums doivent rester dans la fiole.
- Ajouter 100 mL de CDB liquide (étape 1.2) sur le mycélium dans la fiole de chicanerie et compléter le milieu avec 500 mg/L de céfotaxime.
- Incuber encore 4-5 jours dans une boîte sombre à RT sous agitation horizontale continue (agitateur rotatif, 60 tr / min) pour induire une sporulation. Le surnageant deviendra jaunâtre-grisâtre au fur et à mesure que les conidies seront libérées.
- Filtrer une partie (5-10 mL) du liquide contenant des conidies à travers un papier filtre (niveau de rétention des particules de 8-12 μm) dans un tube de collecte stérile de 50 mL. Cela sépare les spores du mycélium.
- Déterminez la concentration de spores à l’aide d’une chambre de comptage cellulaire et d’un microscope. Diluer avec un milieu 1/4 MS sans germes dans de l’eau ultrapure jusqu’à l’obtention des concentrations de spores indiquées ci-dessous.
REMARQUE: Au microscope, les conidies de V. longisporum sont pour la plupart de longues dessinées et de taille 7,1-8,8 μm, tandis que les conidies de V. dahliae sont plus courtes (3,5-5,5 μm) et plutôtsphériques 20. - Utilisez ces conidies fraîchement récoltées comme inoculum. Assurez-vous de mener les expériences toujours avec des conidies fraîchement récoltées et non avec des stocks congelés, car la congélation réduit considérablement le nombre de spores viables19.
- Pour un stockage à long terme, congeler les spores sous forme de solution de spores hautement concentrée (environ 1 x 108 spores/mL) dans du glycérol à 25 % à -80 °C (stockable jusqu’à 1 an). Pour les prochaines expériences, utilisez ces stocks de glycérol pour inoculer le milieu PDB à l’étape 3.2.
4. Un système d’inoculation stérile in vitro à base de boîtes de Pétri
REMARQUE: Pour le système de boîtede Petri 17, assurez-vous que tout l’équipement et les supports sont exempts de germes et que toutes les étapes sont effectuées dans une hotte à flux laminaire.
- Après l’autoclavage, versez le milieu (voir étape 1.3) dans des boîtes de Pétri.
- Après le durcissement du milieu, remballez les boîtes de Petri dans un sac en plastique stérile et conservez-les à l’envers pendant la nuit au réfrigérateur (4-10 °C). Un milieu réfrigéré aide à prévenir le glissement du milieu dans les étapes suivantes.
- Coupez et retirez un canal d’infection et le tiers supérieur du milieu solidifié avec un scalpel (Figure 1A). Évitez d’obtenir du liquide ou de l’air sous le milieu de la gélose pendant la coupe; sinon, le milieu glissera et fermera le canal d’infection.
- Distribuer 50 à 100 graines d’Arabidopsis stérilisées en surface avec une pointe de pipette stérile sur la surface supérieure coupée. Placez les graines dans l’angle où la surface de la gélose coupée entre en contact avec la paroi de la boîte de Pétri afin que les racines puissent pousser entre le milieu et la paroi de la boîte de Pétri. Cela facilitera l’inoculation plus tard.
- Fermez les boîtes de Petri et scellez-les avec du ruban adhésif perméable à l’air pour permettre l’échange de gaz.
- Après stratification pendant 2 jours dans l’obscurité à 4 °C, placez les plaques verticalement dans un rack approprié et faites pousser les plantes à 22 °C ± 1 °C dans des conditions de longue journée (16 h de lumière / 8 h d’obscurité) dans une chambre de croissance.
- Lorsque la majorité des racines atteignent le canal d’infection (environ 9 à 11 jours de semis), posez les plaques horizontalement, ouvrez-les et ajoutez 500 μL de conidies verticillium fraîchement récoltées avec une concentration de 4 x 105 spores / mL directement dans le canal infectieux, en s’assurant que le liquide est réparti uniformément dans le canal.
- De même, préparer les plaques de contrôle en ajoutant 500 μL d’une solution simulée au lieu de spores (milieu 1/4 MS sans germes).
- Incuber les plaques horizontalement pendant quelques minutes jusqu’à ce que le liquide ait trempé et ne puisse pas s’échapper lorsque les plaques sont à nouveau installées verticalement. Ensuite, fermez le couvercle et scellez les plaques avec du ruban adhésif perméable à l’air.
- Incuber les plaques verticalement dans la chambre de croissance. En option, couvrez les parties racinaires avec des boîtes en papier noires pour assombrir les racines et les champignons transmis par le sol (voir19).
- Effectuer les analyses aux points temporels préférés après l’inoculation en fonction de la question de recherche (se référer aux légendes de figure pour les points de temps exacts utilisés ici). Voici quelques suggestions.
- Coupez les feuilles des racines et récoltez les deux séparément. Sortez les bandes de gélose des boîtes de Pétri pour accéder facilement aux racines et retirez-les soigneusement de la gélose à l’aide d’une pince. Congeler immédiatement tout le matériel végétal dans de l’azote liquide.
- Broyer les échantillons dans de l’azote liquide. Extraire l’ADN total de 100 mg de matière foliaire pour déterminer par PCR quantitative (qPCR) la quantité d’ADN fongique par rapport à l’ADN végétal (voir19).
- Broyer les échantillons dans de l’azote liquide. Prendre 100 mg de matériel végétal et extraire l’ARN total. Effectuer une PCR quantitative par transcription inverse (qRT-PCR) pour déterminer l’expression des gènes végétaux (ou gènes fongiques) pendant l’infestation (voir19).
- Retirez soigneusement les racines de la gélose pour éviter toute blessure et examinez-les au microscope fluorescent.
- Déterminer l’induction de gènes marqueurs dans les lignées de rapporteurs végétaux (p. ex. luciférase, β-glucuronidase ou rapporteurs fluorescents 17,19,21).
- Visualiser la propagation fongique à la racine à l’aide de lignes rapporteures fongiques (p. ex., V. longisporum exprimant de manière constitutive la protéine fluorescente verte améliorée, Vl-sGFP9) ou par des techniques de coloration (p. ex., par l’intermédiaire du 5-bromo-4-chloro-3-indoxyl-N-acétyl-bêta-d-glucosaminide (X-bêta-D-Glc-Nac)18).
- Coupez les feuilles des racines et récoltez les deux séparément. Sortez les bandes de gélose des boîtes de Pétri pour accéder facilement aux racines et retirez-les soigneusement de la gélose à l’aide d’une pince. Congeler immédiatement tout le matériel végétal dans de l’azote liquide.
5. Un système d’inoculation stérile in vitro organisé avec des gobelets en plastique
REMARQUE: Comme indiqué dans la première description de cette technique19, assurez-vous que tout l’équipement et les milieux sont exempts de germes et que toutes les étapes sont effectuées dans une hotte à écoulement laminaire.
- Utilisez des gobelets en plastique transparent d’un volume total de 500 mL et stérilisez-les dans un bain d’éthanol à 70% à 75% pendant au moins 20 min. Séchez les tasses dans la hotte à écoulement laminaire.
- Versez le milieu autoclavé (voir étape 1.4) dans les gobelets en plastique. Éventuellement, ajouter le céfotaxime (concentration finale de 50 mg/L) au milieu autoclavé pour éviter les contaminations bactériennes. Utilisez 150 mL de milieu par tasse pour les expériences avec Arabidopsis ou plus de milieu (250-300 mL par tasse) pour les expériences avec des espèces végétales plus grandes (colza, tomate).
- Placer une couche de plastique (stérilisée avant par incubation dans de l’éthanol à 70 % à 75 % pendant 20 min) sur le milieu avant qu’il ne se solidifie (Figure 1B).
REMARQUE: Cette couche de plastique contient quatre trous préfabriqués aux coins pour placer des graines stérilisées en surface. Cela permet aux graines d’accéder au milieu. Plus tard, cette couche de séparation empêche les feuilles de toucher le milieu contenant le champignon, de sorte que les microbes ne peuvent pas attaquer directement les feuilles et doivent emprunter la voie racinaire. Un autre trou est au centre, permettant de couper le canal d’infection. - Lorsque le milieu s’est solidifié, coupez la gélose avec un scalpel à travers le trou central préfabriqué à une profondeur d’environ 1,5 cm. Retirez la gélose coupée pour créer un canal d’infection dans lequel les spores fongiques peuvent être ajoutées plus tard.
- Grattez légèrement le milieu de la gélose avec une pointe de pipette dans les quatre trous plus petits pour interrompre la peau solidifiée (cela permet aux graines d’absorber l’eau du milieu aqueux de la gélose). Placez les graines à l’aide d’une pointe de pipette dans les trous plus petits.
- Fermez le gobelet en plastique avec un deuxième gobelet en plastique inversé et scellez-le avec du ruban adhésif perméable à l’air. Le ruban doit permettre l’échange de gaz.
- Après stratification pendant 3 jours dans l’obscurité à 4 °C, incuber les systèmes de gobelets sous 12 h de lumière / 12 h d’obscurité (Arabidopsis, colza) ou 16 h de lumière / 8 h d’obscurité (tomate) dans des chambres de croissance à une température constante de 22 °C et 60% d’humidité.
- Suivez l’âge recommandé des plantes pour l’inoculation: 21 jours pour Arabidopsis; 5-7 jours pour le colza; 12 jours pour la tomate.
- Inoculez les plantules avec Verticillium en ajoutant 1 mL de solution de conidies (concentration recommandée : 4 x 105 spores/mL) dans le canal infectieux. Pour préparer les échantillons témoins, ajouter 1 mL de solution simulée sans spores (milieu 1/4 MS sans germes) dans le canal.
- Effectuer les analyses aux points temporels préférés après l’inoculation en fonction de la question de recherche (se référer aux légendes de figure pour les points de temps exacts utilisés ici). Voici quelques suggestions.
- Prenez des photos des plantes avec un appareil photo numérique d’en haut en gardant la même distance pour chaque photo. Quantifier la surface foliaire (p. ex., avec ImageJ22 ou BlattFlaeche17,19; utiliser la longueur des tasses pour définir l’échelle) et comparer les groupes infectés et témoins. Catégoriser le développement des symptômes de la maladie (p. ex., feuilles plus petites, plus jaunâtres ou nécrotiques).
REMARQUE: S’il y a des tiges sur Arabidopsis, retirez-les pour obtenir de meilleures photos des rosettes. - Enlever les racines et définir la biomasse (poids frais) des pousses des échantillons infectés et témoins en les pesant. Déterminer le poids relativement frais19.
- Prélever les échantillons pour les analyses moléculaires comme suit.
- Arabidopsis: Enlevez les tiges s’il y en a. Coupez les rosettes à la base des racines. Assurez-vous d’exclure tout le matériel racinaire de l’échantillon et de récolter des rosettes entières. Combinez 4-5 rosettes de différentes plantes en un seul échantillon et congelez la matière foliaire dans de l’azote liquide.
- Retirez soigneusement les racines du milieu avec des pinces, pressez-les et tamponnez-les avec une serviette en papier pour enlever les restes de gélose et combinez 4 à 5 racines de différentes plantes en un seul échantillon. Congeler immédiatement dans de l’azote liquide.
- Colza/tomate : Couper les segments de tige de l’hypocotyle (p. ex., 1 cm de longueur; toujours prendre la même région de tige). Mélanger le matériel de 4-5 plantes dans chaque échantillon et congeler dans de l’azote liquide.
- Broyer les échantillons dans de l’azote liquide. Extraire l’ADN total de 100 mg de la feuille ou de la tige pour déterminer par qPCR la quantité d’ADN fongique par rapport à l’ADN de la plante (voir19).
- Broyer les échantillons dans de l’azote liquide, prélever 100 mg de matériel végétal et extraire l’ARN total. Effectuer une qRT-PCR pour déterminer l’expression des gènes végétaux (ou gènes fongiques) lors de l’infestation (voir19).
- Prenez des photos des plantes avec un appareil photo numérique d’en haut en gardant la même distance pour chaque photo. Quantifier la surface foliaire (p. ex., avec ImageJ22 ou BlattFlaeche17,19; utiliser la longueur des tasses pour définir l’échelle) et comparer les groupes infectés et témoins. Catégoriser le développement des symptômes de la maladie (p. ex., feuilles plus petites, plus jaunâtres ou nécrotiques).
6. Un système d’inoculation à base de sol dans des pots
- Mélangez soigneusement le sol et le sable dans un rapport volumétrique de 3:1 (sol:sable) pour faciliter le lavage du substrat des racines. Versez le mélange dans un sac autoclave. Si le mélange est trop sec, ajoutez une quantité appropriée d’eau et mélangez-la dans le substrat. Cuire à la vapeur à 80 °C pendant 20 min dans un autoclave pour minimiser les contaminations microbiennes.
REMARQUE: Évitez de chauffer à plus de 80 ° C, car cela pourrait affecter les nutriments organiques du sol. - Remplissez les pots avec le mélange terre-sable et transférez-les dans des plateaux. Ajoutez de l’eau dans les plateaux à environ 1/3 de la hauteur d’un pot, de sorte que le mélange sol-sable soit bien trempé dans l’eau. De plus, vaporisez le substrat avec un flacon pulvérisateur pour assurer des conditions de départ humides.
- Semez 3 à 4 graines dans chaque pot (Figure 1C) en vous assurant que les graines sont suffisamment éloignées les unes des autres. Conservez-les pendant 3 jours dans l’obscurité à 4 °C pour la stratification afin de synchroniser la germination.
REMARQUE: Pré-cultiver un excès de plantes, ce qui permet une sélection de plantes de taille similaire pour les expériences d’inoculation et réduit les écarts dus aux différences individuelles. - Laissez les semis pousser dans des conditions de longue journée (16 h de lumière / 8 h d’obscurité; température constante de 22 ° C; 60% d’humidité) avec un arrosage régulier.
- Suivez l’âge recommandé des plantes pour l’inoculation: 21 jours pour Arabidopsis, 7 jours pour le colza et 10 jours pour la tomate. Cueillez des plantes de taille similaire pour effectuer l’inoculation par immersion racinaire 15,17,23,24. Retirez la terre des pots et excusez soigneusement les racines.
- Ne lavez doucement que les racines dans un récipient d’eau et gardez les rosettes hors de l’eau. Incuber les racines lavées pendant 60 min dans une boîte de Petri contenant la solution de spores de Verticillium (concentration recommandée : 2 x 106 spores/mL). Pour le groupe témoin non infecté, incuber les racines pendant 60 min dans la solution simulée sans spores (milieu 1/4 MS sans germes).
- Préparez de nouveaux pots avec un sol humide et stérilisé à la vapeur (80 °C pendant 20 min) sans sable. Utilisez une pointe de pipette pour faire un trou au centre du sol dans chaque pot.
- Placez directement les racines dans le trou (ne transférez qu’une seule plante par pot). Après avoir inséré les racines, assurez-vous de remplir soigneusement les trous avec de la terre. Évitez d’appuyer sur le sol, sinon le rempotage peut causer des symptômes de stress tels que des feuilles violettes.
- Cultivez des groupes infectés et témoins dans des conditions de longue journée (16 h de lumière / 8 h d’obscurité; une température constante de 22 ° C; 60% d’humidité) avec un arrosage régulier.
- Effectuer les analyses aux points temporels préférés après l’inoculation en fonction de la question de recherche (se référer aux légendes de figure pour les points de temps exacts utilisés ici). Voici quelques suggestions.
- Prenez des photos des plantes avec un appareil photo numérique d’en haut, en gardant la même distance pour chaque photo. Quantifier la surface foliaire (p. ex., avec ImageJ22 ou BlattFlaeche17,19; utiliser le diamètre des pots pour définir l’échelle) et comparer les groupes infectés et témoins. Catégoriser le développement des symptômes de la maladie (p. ex., feuilles plus petites, plus jaunâtres ou nécrotiques)13.
REMARQUE: Enlever les tiges d’Arabidopsis facilite la prise de photos des rosettes. - Enlever les racines et définir la biomasse (poids frais) des pousses des échantillons infectés et témoins en les pesant. Déterminer le poids relativement frais19.
- Alternativement, mesurez la hauteur de la plante ou catégorisez l’excroissance fongique des segments de tige pour évaluer la gravité de la maladie13.
- Prélever des échantillons pour des analyses moléculaires comme suit.
- Arabidopsis: Enlevez les tiges. Coupez les rosettes au niveau de la couronne racinaire. Combinez 4-5 rosettes de différentes plantes en un seul échantillon. Congeler la matière foliaire dans de l’azote liquide.
REMARQUE: Dans le cas des racines, il est difficile de les nettoyer suffisamment du sol sans reprogrammer l’expression des gènes par lavage. - Colza/tomate : Couper les segments de tige de l’hypocotyle (p. ex., 1 cm de longueur; toujours prendre la même région de tige). Combinez le matériel de 4 à 5 plantes en un seul échantillon et congelez-le dans de l’azote liquide.
- Broyer les échantillons dans de l’azote liquide. Extraire l’ADN total de 100 mg de feuille ou de tige pour déterminer par qPCR la quantité d’ADN fongique par rapport à l’ADN végétal (voir19).
- Broyer les échantillons dans de l’azote liquide, prélever 100 mg de matériel végétal et extraire l’ARN total. Effectuer une qRT-PCR pour déterminer l’expression des gènes végétaux (ou gènes fongiques) lors de l’infestation (voir19).
- Prenez des photos des plantes avec un appareil photo numérique d’en haut, en gardant la même distance pour chaque photo. Quantifier la surface foliaire (p. ex., avec ImageJ22 ou BlattFlaeche17,19; utiliser le diamètre des pots pour définir l’échelle) et comparer les groupes infectés et témoins. Catégoriser le développement des symptômes de la maladie (p. ex., feuilles plus petites, plus jaunâtres ou nécrotiques)13.
7. Analyse des données
- Calculer la moyenne et l’écart-type (± SD) en fonction des répliques biologiques.
- Calculez les valeurs relatives en divisant tous les résultats du groupe infecté par le résultat du contrôle. Affichez la moyenne comme, par exemple, « par rapport à la simulation » ou « par rapport au type sauvage ».
- Déterminer la signification statistique entre les groupes.

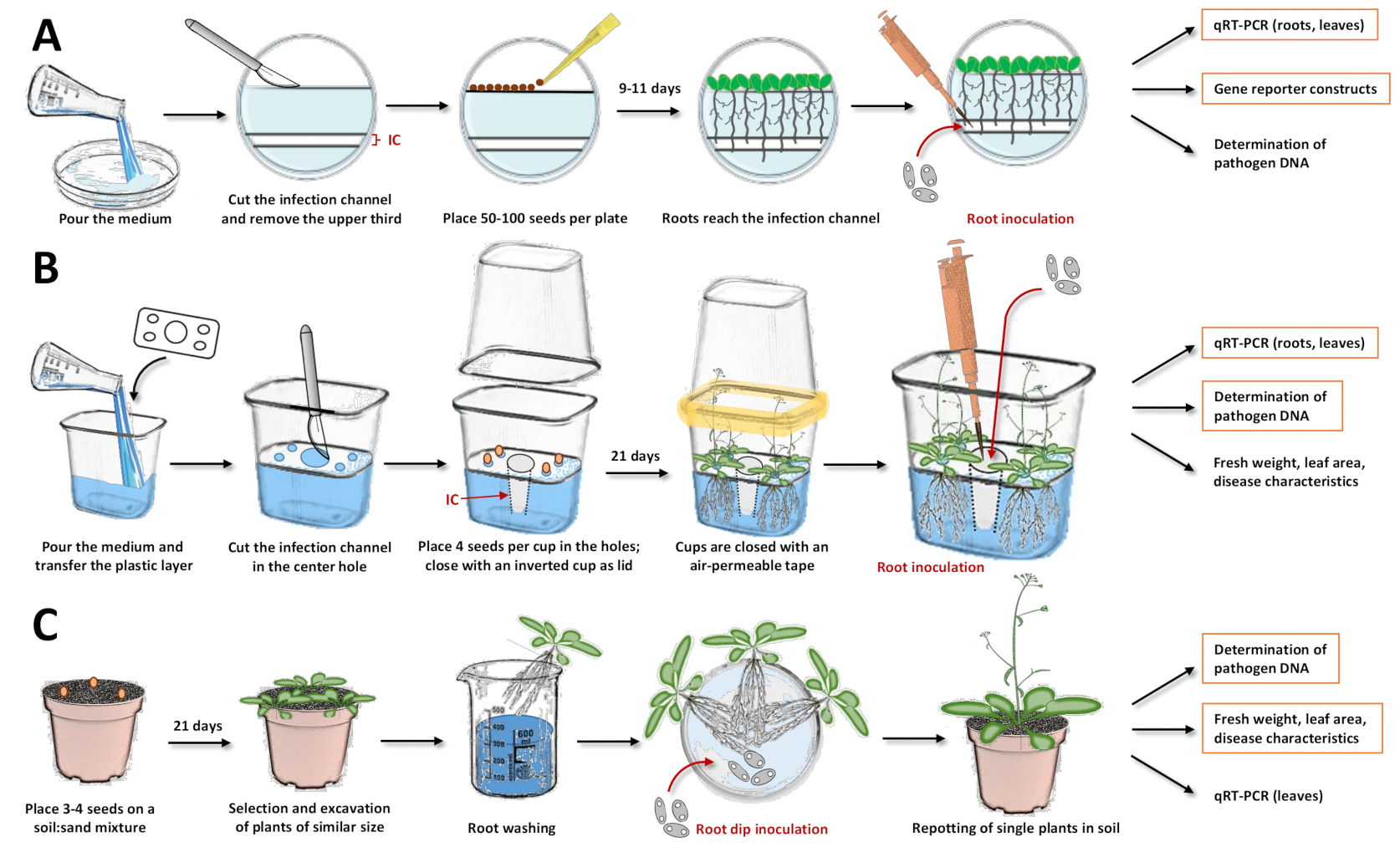
Figure 1 : Compilation des trois systèmes d’inoculation et des différentes étapes des protocoles. Ces figures illustrent les systèmes avec la plante modèle Arabidopsis thaliana. Pour les autres espèces végétales, le moment doit être ajusté. Les cases orange sont en surbrillance, pour lesquelles des analyses ultérieures sont les plus recommandées avec le système respectif. (A) Pour le système d’inoculation dans les boîtes de Pétri17, versez le milieu et laissez-le se solidifier. Conservez les assiettes au réfrigérateur pendant la nuit. Ensuite, coupez et retirez le tiers supérieur ainsi que le canal d’infection (CI) avec un scalpel (les zones blanches de l’illustration ont été retirées de la gélose, tandis que les zones bleuâtres représentent la gélose). Placez les graines sur la surface coupée et fermez les boîtes de Pétri. Après stratification, placez les plaques verticalement et laissez les plantes pousser. Une fois que la plupart des racines ont atteint le canal d’infection, ajoutez la solution de spores avec une pipette directement dans le canal. Assurez-vous que la solution est répartie uniformément. Fermez les boîtes de Petri et incuvrez-les verticalement dans une chambre de croissance. Les approches qui peuvent suivre sont l’analyse expressionnelle avec PCR quantitative à transcription inverse (qRT-PCR), la microscopie avec des lignes rapporteures et la quantification de l’ADN microbien. (B) Pour le système d’inoculation dans les gobelets en plastique19, versez le milieu et transférez la couche de plastique de séparation avec les trous préfabriqués (quatre petits trous dans les coins pour placer les graines et un grand trou au centre pour le canal d’infection). Laissez le milieu se solidifier. Coupez et retirez le milieu de la gélose dans le trou central avec un scalpel pour obtenir le canal d’infection (CI). Grattez le milieu dans les petits trous et transférez les graines. Fermez la tasse avec une tasse inversée et scellez-la avec du ruban adhésif perméable à l’air (symbolisé en jaune). Laissez les plantes pousser. Pour l’inoculation, ajouter la solution de spores avec une pipette directement dans le canal d’infection. Fermez le système et continuez la culture dans la chambre de croissance. Les approches qui peuvent suivre sont l’analyse expressionnelle avec qRT-PCR, la quantification de l’ADN microbien et la détermination du poids frais, de la surface foliaire ou d’autres caractéristiques de la maladie. (C) « Inoculation par immersion racinaire"15,17,23,24: pour le système d’inoculation à base de sol, remplir les pots avec un mélange sol/sable. Transférez les graines et laissez les semis pousser. Excaver des plantes de taille similaire et laver les racines dans l’eau. Placez les racines lavées dans une boîte de Pétri contenant la solution avec les spores. Après l’incubation, insérez des plantes individuelles dans des pots avec de la terre. Les approches qui peuvent suivre sont l’analyse expressionnelle dans les feuilles avec qRT-PCR, la quantification de l’ADN microbien et la détermination du poids frais, de la surface foliaire ou d’autres caractéristiques de la maladie. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
Conformément au protocole, les plantes ont été cultivées et inoculées avec V. longisporum (souche Vl4325) ou V. dahliae (isolat JR218). Divers scénarios ont été conçus pour prouver l’efficacité et mettre en évidence certaines capacités des protocoles donnés. Des résultats représentatifs sont présentés.
L’induction expressionnelle des gènes impliqués dans la biosynthèse antimicrobienne indol-gluco...
Discussion
En raison des énormes pertes de rendement causées par les phytopathogènes transmis par le sol1, une amélioration des stratégies agricoles ou des variétés de cultures est nécessaire. La compréhension limitée de la pathogenèse des maladies transmises par le sol entrave le développement de plantes plus résistantes. Les mécanismes pathologiques sous-jacents doivent être explorés, pour lesquels une plate-forme méthodologique robuste est nécessaire. Les procédures d’inoculation rapp...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Les auteurs remercient Tim Iven et Jaqueline Komorek pour leurs travaux antérieurs sur ces méthodes, le groupe de Wolfgang Dröge-Laser (Département de biologie pharmaceutique, Université de Würzburg, Allemagne) pour avoir fourni l’équipement et les ressources nécessaires à ce travail, et Wolfgang Dröge-Laser ainsi que Philipp Kreisz (tous deux de l’Université de Würzburg) pour la relecture critique du manuscrit. Cette étude a été soutenue par la « Deutsche Forschungsgemeinschaft » (DFG, DR273/15-1,2).
matériels
| Name | Company | Catalog Number | Comments |
| Agar (Gelrite) | Carl Roth | Nr. 0039 | all systems described require Gelrite |
| Arabidopsis thaliana wild-type | NASC stock | Col-0 (N1092) | |
| Autoclave | Systec | VE-100 | |
| BlattFlaeche | Datinf GmbH | BlattFlaeche | software to determine leaf areas |
| Brassica napus wild-type | see Floerl et al., 2008 | rapid-cycling rape | genome ACaacc |
| Cefotaxime sodium | Duchefa | C0111 | |
| Chicanery flask 500 mL | Duran Group / neoLab | E-1090 | Erlenmeyer flask with four baffles |
| Collection tubes 50 mL | Sarstedt | 62.547.254 | 114 x 28 mm |
| Czapek Dextrose Broth medium | Duchefa | C1714 | |
| Digital camera | Nikon | D3100 18-55 VR | |
| Exsiccator (Desiccator ) | Duran Group | 200 DN, 5.8 L | Seal with lid to hold chlorine gas |
| Fluorescence Microscope | Leica | Leica TCS SP5 II | |
| HCl | Carl Roth | P074.3 | |
| KNO3 | Carl Roth | P021.1 | ≥ 99 % |
| KOH | Carl Roth | 6751 | |
| Leukopor | BSN medical GmbH | 2454-00 AP | non-woven tape 2.5 cm x 9.2 m |
| MES (2-(N-morpholino)ethanesulfonic acid) | Carl Roth | 4256.2 | Pufferan ≥ 99 % |
| MgSO4 | Carl Roth | T888.1 | Magnesiumsulfate-Heptahydrate |
| Murashige & Skoog medium (MS) | Duchefa | M0222 | MS including vitamins |
| NaClO | Carl Roth | 9062.1 | |
| Percival growth chambers | CLF Plant Climatics GmbH | AR-66L2 | |
| Petri-dishes | Sarstedt | 82.1473.001 | size ØxH: 92 × 16 mm |
| Plastic cups (500 mL, transparent) | Pro-pac, salad boxx | 5070 | size: 108 × 81 × 102 mm |
| Pleated cellulose filter | Hartenstein | FF12 | particle retention level 8–12 μm |
| poly klima growth chamber | poly klima GmbH | PK 520 WLED | |
| Potato Dextrose Broth medium | SIGMA Aldrich | P6685 | for microbiology |
| Pots | Pöppelmann GmbH | TO 7 D or TO 9,5 D | Ø 7 cm resp. Ø 9.5 cm |
| PromMYB51::YFP | see Poncini et al., 2017 | MYB51 reporter line | YFP (i.e. 3xmVenus with NLS) |
| Reaction tubes 2 mL | Sarstedt | 72.695.400 | PCR Performance tested |
| Rotary (orbital) shaker | Edmund Bühler | SM 30 C control | |
| Sand (bird sand) | Pet Bistro, Müller Holding | 786157 | |
| Soil | Einheitserde spezial | SP Pikier (SP ED 63 P) | |
| Solanum lycopersicum wild-type | see Chavarro-Carrero et al., 2021 | Type: Moneymaker | |
| Thoma cell counting chamber | Marienfeld | 642710 | depth 0.020 mm; 0.0025 mm2 |
| Ultrapure water (Milli-Q purified water) | MERK | IQ 7003/7005 | water obtained after purification |
| Verticillium dahliae | see Reusche et al., 2014 | isolate JR2 | |
| Verticillium longisporum | Zeise and von Tiedemann, 2002 | strain Vl43 |
Références
- Mendes, R., Garbeva, P., Raaijmakers, J. M. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiology Review. 37 (5), 634-663 (2013).
- Yadeta, K. A., Thomma, B. P. H. J. The xylem as battleground for plant hosts and vascular wilt pathogens. Frontiers in Plant Science. 4, 97 (2013).
- Delgado-Baquerizo, M., et al. The proportion of soil-borne pathogens increases with warming at the global scale. Nature Climate Change. 10 (6), 550-554 (2020).
- Berendsen, R. L., et al. Disease-induced assemblage of a plant-beneficial bacterial consortium. The ISME Journal. 12 (6), 1496-1507 (2018).
- Yuan, J., et al. Root exudates drive the soil-borne legacy of aboveground pathogen infection. Microbiome. 6 (1), 156 (2018).
- Liu, H., et al. Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytologist. 229 (5), 2873-2885 (2021).
- Wang, H., Liu, R., You, M. P., Barbetti, M. J., Chen, Y. Pathogen biocontrol using plant growth-promoting bacteria (PGPR): role of bacterial diversity. Microorganisms. 9 (9), 1988 (2021).
- Inderbitzin, P., Subbarao, K. V. Verticillium systematics and evolution: how confusion impedes Verticillium wilt management and how to resolve it. Phytopathology. 104 (6), 564-574 (2014).
- Eynck, C., Koopmann, B., Grunewaldt-Stoecker, G., Karlowsky, P., von Tiedemann, A. Differential interactions of Verticillium longisporum und V. dahliae with Brassica napus with molecular and histological techniques. European Journal of Plant Pathology. 118 (3), 259-274 (2007).
- Floerl, S., et al. Defence reactions in the apoplastic proteome of oilseed rape (Brassica napus var. napus) attenuate Verticillium longisporum growth but not disease symptoms. BMC Plant Biology. 8, 129 (2008).
- Leonard, M., et al. Verticillium longisporum elicits media-dependent secretome responses with capacity to distinguish between plant-related environments. Frontiers in Microbiology. 11, 1876 (2020).
- Depotter, J. R. L., et al. Verticillium longisporum, the invisible threat to oilseed rape and other brassicaceous plant hosts. Molecular Plant Pathology. 17 (7), 1004-1016 (2016).
- Fröschel, C., et al. A gain-of-function screen reveals redundant ERF transcription factors providing opportunities for resistance breeding toward the vascular fungal pathogen Verticillium longisporum. Molecular Plant-Microbe Interactions. 32 (9), 1095-1109 (2019).
- Zhou, L., Hu, Q., Johansson, A., Dixelius, C. Verticillium longisporum and V. dahliae: infection and disease in Brassica napus. Plant Pathology. 55 (1), 137-144 (2006).
- Ralhan, A., et al. The vascular pathogen Verticillium longisporum requires a jasmonic acid-independent COI1 function in roots to elicit disease symptoms in Arabidopsis shoots. Plant Physiology. 159 (3), 1192-1203 (2012).
- Reusche, M., et al. Stabilization of cytokinin levels enhances Arabidopsis resistance against Verticillium longisporum. Molecular Plant-Microbe Interactions. 26 (8), 850-860 (2013).
- Iven, T., et al. Transcriptional activation and production of tryptophan-derived secondary metabolites in Arabidopsis roots contributes to the defense against the fungal vascular pathogen Verticillium longisporum. Molecular Plant. 5 (6), 1389-1402 (2012).
- Reusche, M., et al. Infections with the vascular pathogens Verticillium longisporum and Verticillium dahliae induce distinct disease symptoms and differentially affect drought stress tolerance of Arabidopsis thaliana. Environmental and Experimental Botany. 108, 23-37 (2014).
- Fröschel, C. In-depth evaluation of root infection systems using the vascular fungus Verticillium longisporum as soil-borne model pathogen. Plant Methods. 17 (1), 57 (2021).
- Karapapa, V. K., Bainbridge, B. W., Heale, J. B. Morphological and molecular characterization of Verticillium longisporum comb, nov., pathogenic to oilseed rape. Mycological Research. 101 (11), 1281-1294 (1997).
- Poncini, L., et al. In roots of Arabidopsis thaliana, the damage-associated molecular pattern AtPep1 is a stronger elicitor of immune signalling than flg22 or the chitin heptamer. PLoS One. 12 (10), 1-21 (2017).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 9 (7), 671-675 (2012).
- Fradin, E. F., et al. Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiology. 150 (1), 320-332 (2009).
- Singh, S., et al. The plant host Brassica napus induces in the pathogen Verticillium longisporum the expression of functional catalase peroxidase which is required for the late phase of disease. Molecular Plant-Microbe Interactions. 25 (4), 569-581 (2012).
- Zeise, K., von Tiedemann, A. Application of RAPD-PCR for virulence type analysis within Verticillium dahliae and Verticillium longisporum. Journal of Phytopathology. 150 (10), 557-563 (2002).
- Fröschel, C., et al. Plant roots employ cell-layer-specific programs to respond to pathogenic and beneficial microbes. Cell Host & Microbe. 29 (2), 299-310 (2021).
- Gigolashvili, T., et al. The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. The Plant Journal. 50 (5), 886-901 (2007).
- Back, M. A., Haydock, P. P. J., Jenkinson, P. Disease complexes involving plant parasitic nematodes and soilborne pathogens. Plant Pathology. 51 (6), 683-697 (2002).
- Behrens, F. H., et al. Suppression of abscisic acid biosynthesis at the early infection stage of Verticillium longisporum in oilseed rape (Brassica napus). Molecular Plant Pathology. 20 (12), 1645-1661 (2019).
- Vorholt, J. A., Vogel, C., Carlström, C. I., Müller, D. B. Establishing causality: opportunities of synthetic communities for plant microbiome research. Cell Host & Microbe. 22 (2), 142-155 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.