
Method Article
A Guide to Examining Intramuscular Fat Formation and its Cellular Origin in Skeletal Muscle
In This Article
Summary
The replacement of healthy muscle tissue with intramuscular fat is a prominent feature of human diseases and conditions. This protocol outlines how to visualize, image, and quantify intramuscular fat, allowing the rigorous study of the mechanisms underlying intramuscular fat formation.
Abstract
Fibro-adipogenic progenitors (FAPs) are mesenchymal stromal cells that play a crucial role during skeletal muscle homeostasis and regeneration. FAPs build and maintain the extracellular matrix that acts as a molecular myofiber scaffold. In addition, FAPs are indispensable for myofiber regeneration as they secrete a multitude of beneficial factors sensed by the muscle stem cells (MuSCs). In diseased states, however, FAPs are the cellular origin of intramuscular fat and fibrotic scar tissue. This fatty fibrosis is a hallmark of sarcopenia and neuromuscular diseases, such as Duchenne Muscular Dystrophy. One significant barrier in determining why and how FAPs differentiate into intramuscular fat is effective preservation and subsequent visualization of adipocytes, especially in frozen tissue sections. Conventional methods of skeletal muscle tissue processing, such as snap-freezing, do not properly preserve the morphology of individual adipocytes, thereby preventing accurate visualization and quantification. To overcome this hurdle, a rigorous protocol was developed that preserves adipocyte morphology in skeletal muscle sections allowing visualization, imaging, and quantification of intramuscular fat. The protocol also outlines how to process a portion of muscle tissue for RT-qPCR, enabling users to confirm observed changes in fat formation by viewing differences in the expression of adipogenic genes. Additionally, it can be adapted to visualize adipocytes by whole-mount immunofluorescence of muscle samples. Finally, this protocol outlines how to perform genetic lineage tracing of Pdgfrα-expressing FAPs to study the adipogenic conversion of FAPs. This protocol consistently yields high-resolution and morphologically accurate immunofluorescent images of adipocytes, along with confirmation by RT-qPCR, allowing for robust, rigorous, and reproducible visualization and quantification of intramuscular fat. Together, the analysis pipeline described here is the first step to improving our understanding of how FAPs differentiate into intramuscular fat, and provides a framework to validate novel interventions to prevent fat formation.
Introduction
The infiltration of healthy muscle tissue with fatty fibrosis is a prominent feature of Duchenne Muscular Dystrophy (DMD) and other neuromuscular diseases, as well as sarcopenia, obesity, and diabetes1,2,3,4,5,6,7,8,9,10. Although increased fat infiltration in these conditions is strongly associated with decreased muscle function, our knowledge of why and how intramuscular fat forms is still limited. FAPs are a multipotent mesenchymal stromal cell population present in most adult organs, including skeletal muscle11,12. With age and in chronic diseases, however, FAPs produce fibrotic scar tissue and differentiate into adipocytes, which are located between individual myofibers and form intramuscular fat13,14,15,16,17,18,19,20.
To start combating intramuscular fat formation, the mechanisms of how FAPs turn into adipocytes need to be defined. PDGFRα is the "gold-standard" marker in the field to identify FAPs within the muscle of multiple species13,16,17,18,20,21,22,23,24,25,26,27. As a result, several murine tamoxifen-inducible Cre lines, under the control of the Pdgfrα promotor, have been generated, allowing for genetically manipulating FAPs in vivo using the Cre-LoxP system27,28,29. For example, by combining this inducible Cre line with a genetic reporter, lineage tracing of FAPs can be performed, a strategy we have successfully applied to fate map FAPs in muscle and white adipose tissue20,30. Besides lineage tracing, these Cre lines provide valuable tools to study the FAP-to-fat conversion.
One major obstacle in defining the mechanism of the adipogenic conversion of FAPs into intramuscular fat is the ability to rigorously and reproducibly quantify the amount of intramuscular fat that has formed under different conditions. The key is to balance the preservation of muscle and fat tissue and match this with the available staining methods to visualize adipocytes. For example, skeletal muscle is often snap-frozen without prior fixation, preserving myofibers but disrupting adipocyte morphology (Figure 1). In contrast, fixation followed by paraffin embedding, while displaying the best tissue histology, including adipocytes, removes all lipids, thereby rendering most lipophilic dyes, such as the commonly used dye Oil Red O, unusable.

Figure 1: Representative images of intramuscular fat in snap-frozen versus fixed muscle tissues. (A) Schematic overview of the experimental setup. Immunofluorescent images showing adipocytes (yellow), myofibers (gray), and nuclei (cyan) within both (B) snap-frozen and (C) fixed TAs at 21 days post glycerol injury. Scale bars: 50 µm. Please click here to view a larger version of this figure.
{kind=link}
The protocol described here preserves myofiber and adipocyte morphology and allows visualization, and analysis, of multiple cell types. This approach is based on immunofluorescence staining of adipocytes in paraformaldehyde (PFA)-fixed muscle tissue, which allows for co-staining with multiple antibodies. It can also be easily adapted to spatially display intramuscular fat in intact tissue using whole-mount imaging, thereby providing information on the cellular microenvironment of fat within the muscle. In addition, this protocol can be combined with our recently published approach to determine the cross-sectional area of myofibers in fixed muscle tissues31, an important measurement to assess muscle health. Combining this approach with genetic lineage tracing to fate-map the differentiation of FAPs into adipocytes is also outlined here. Thus, the versatile protocol described here enables rigorous and reproducible assessment of FAPs and their differentiation into intramuscular fat in tissue sections and intact tissues.

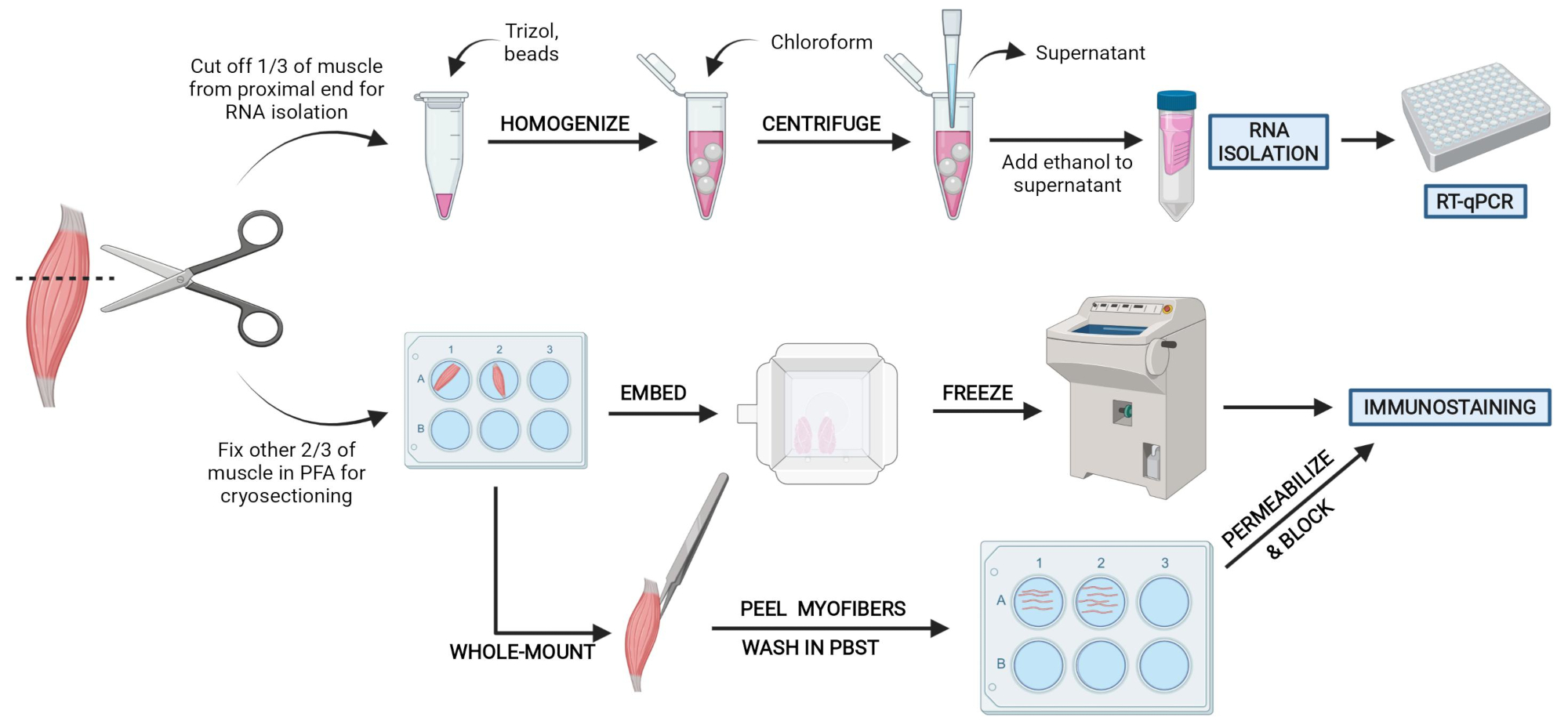
Figure 2: Schematic protocol overview. Schematic overview of tissue processing in which one-third of the TA is removed, snap-frozen, and homogenized for subsequent RNA isolation and transcription analysis via RT-qPCR. The other two-thirds of the TA is PFA-fixed and processed for immunostaining on frozen sections or whole-mount fibers. Please click here to view a larger version of this figure.
{kind=link}
Protocol
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Florida.
1. Genetic lineage tracing of FAPs
NOTE: If genetic lineage tracing of FAPs is not desired, step 1 can be skipped.
- To perform lineage tracing of FAPs, obtain the necessary mouse alleles.
NOTE: Several tamoxifen-inducible Cre lines, under the control of the Pdgfrα promotor, have been generated to successfully target FAPs, including from the Hogan29, Rando27, and Bergles32 laboratories. As a genetic reporter of Cre activity, numerous Rosa26 reporter alleles are available such as Rosa26EYFP33. While it is suggested for each laboratory to determine which Cre-Reporter combination is most effective, by crossing PdgfrαCreERT2 mice (29 and Jax# 032770) to the Rosa26EYFP (33 and Jax# 006148) reporter, the resulting PdgfrαCreERT2; Rosa26EYFPmice can be used to efficiently and specifically mark FAPs20. To trace the fate of mature FAPs, it is recommended to wait until mice have reached at least ~10 weeks of age before administering tamoxifen. Lineage tracing experiments can be performed on both males and females. - Tamoxifen administration through oral gavaging
- Prepare 40 mg/mL tamoxifen in corn oil and vortex well to mix 1 day prior to gavaging. Incubate O/N at 37 °C in a rotating hybridization oven.
CAUTION: Tamoxifen is a carcinogen and should be handled carefully. Always wear gloves when handling and wear a mask when weighing it as a powder, as there is a danger of inhalation. - Clean the area according to protocol and attach a gavaging needle to a 1 mL syringe. Draw 200 µL of tamoxifen into the syringe.
- Scruff PdgfrαCreERT2 ; Rosa26EYFP mice (10 weeks old; both sexes used) by placing them on a flat surface and firmly gripping the base of the tail. Use a free hand to grasp the middle of the mouse with the thumb and index finger, then gently and with slight pressure slide the grip until just past the shoulders.
- Pinch the skin back with the thumb and index finger, pick up the mouse and flip the hand so that the mouse is facing the user, and tuck the tail between the pinky and ring finger of the hand holding the mouse.
- At this point, ensure that the mouse is well immobilized and unable to move its head or arms. Insert the gavaging needle into the mouth and use it to slightly tilt the head of the mouse back; this allows the esophagus to be better accessible.
- Carefully and slowly insert the needle into the esophagus. Do not force the needle if any resistance is met; the needle should slide down easily. Slowly inject the tamoxifen. Monitor the mice for 15-20 min to ensure no problems occur while gavaging.
NOTE: Administration of tamoxifen on 2 consecutive days typically results in ~75%-85% recombination efficiency of FAPs without causing any adverse effects. It is recommended that the user waits for 1-2 weeks before inducing injury, which will allow the remaining tamoxifen to be removed from the system and any remaining protein to be turned over.
- Prepare 40 mg/mL tamoxifen in corn oil and vortex well to mix 1 day prior to gavaging. Incubate O/N at 37 °C in a rotating hybridization oven.
2. Injury of tibialis anterior (TA) muscle
NOTE: To study intramuscular fat, it is recommended to use a glycerol-based injury model (50% glycerol in sterile saline), which results in massive intramuscular fat formation34,35,36,37.
- Prepare the anesthetic machine by adding isoflurane and ensuring the tubes to both the mouse chamber and nose cone are open. Clean the chamber and work area with either 70% ethanol or peroxide solution (depending on protocols).
- Set the oxygen flow rate to 2.5 L/min and the isoflurane concentration to 2.5%. Place a mouse into the anesthesia chamber and wait ~5 min for it to be anesthetized.
- Place the mouse down supine on a clean heating pad and insert the nose into the nose cone. Gently apply vet ophthalmic ointment to the eyes with a cotton-tipped applicator to prevent dryness while under anesthesia. Continuously monitor anesthesia and perform a toe pinch on the mouse prior to the injury to ensure the mouse is fully anesthetized.
- Clean the leg to be injected with a fresh alcohol wipe to disinfect.
- Draw up 30-50 µL of 50% glycerol (depending on the size of the mice) into the insulin syringe. Gently brush up the hair on the shin to expose the location of the TA.
NOTE: It is easier to move the hair and achieve better visualization when it is still wet from the alcohol wipe. - After locating the TA (just lateral to the tibia, it slightly protrudes through the skin and can be felt with gentle palpation), insert the needle into the TA distally, near the ankle. Fully insert the needle into the muscle and slowly inject glycerol while gradually withdrawing the needle, which helps to injure most of the muscle.
NOTE: It is best to insert the needle parallel to the leg, with just a slightly elevated angle. A good injury typically causes dorsiflexion as the TA contracts after withdrawing the needle. If the mouse's toes spread out, it is likely that the extensor digitorum longus (EDL) muscle was injected. - Place the mouse back in the cage and monitor for about 15-20 min to ensure anesthesia recovery.
- Discard the needle in a sharps container. Never recap a needle.
NOTE: Analgesia post glycerol injection should be provided as approved by the Institutional Animal Care and Use Committee. Adipocytes can be observed as soon as 5 days post-injury (dpi). By 7 dpi, all adipocytes have formed, and by 21 dpi, they have fully matured.
3. Tissue harvest
- Prepare 4% PFA in 1x PBS and place on ice prior to starting harvesting.
- Place any plates (12- or 24-well) being used for muscle fixation on ice and add 4% PFA to each well, making sure that each well has 10-20 times more volume of PFA than the tissue being fixed.
- Between 7 and 21 days post glycerol injury, euthanize the mouse according to Institutional guidelines (i.e., isoflurane overdose followed by cervical dislocation).
- Begin harvesting any tissues to be used for histology or RNA isolation.
NOTE: Tissues should be snap-frozen or placed in PFA within 10-15 min of sacrificing (Figure 2). - Liberally spray any areas of the mouse to be cut into with 70% ethanol to help keep hair off dissecting area and instruments.
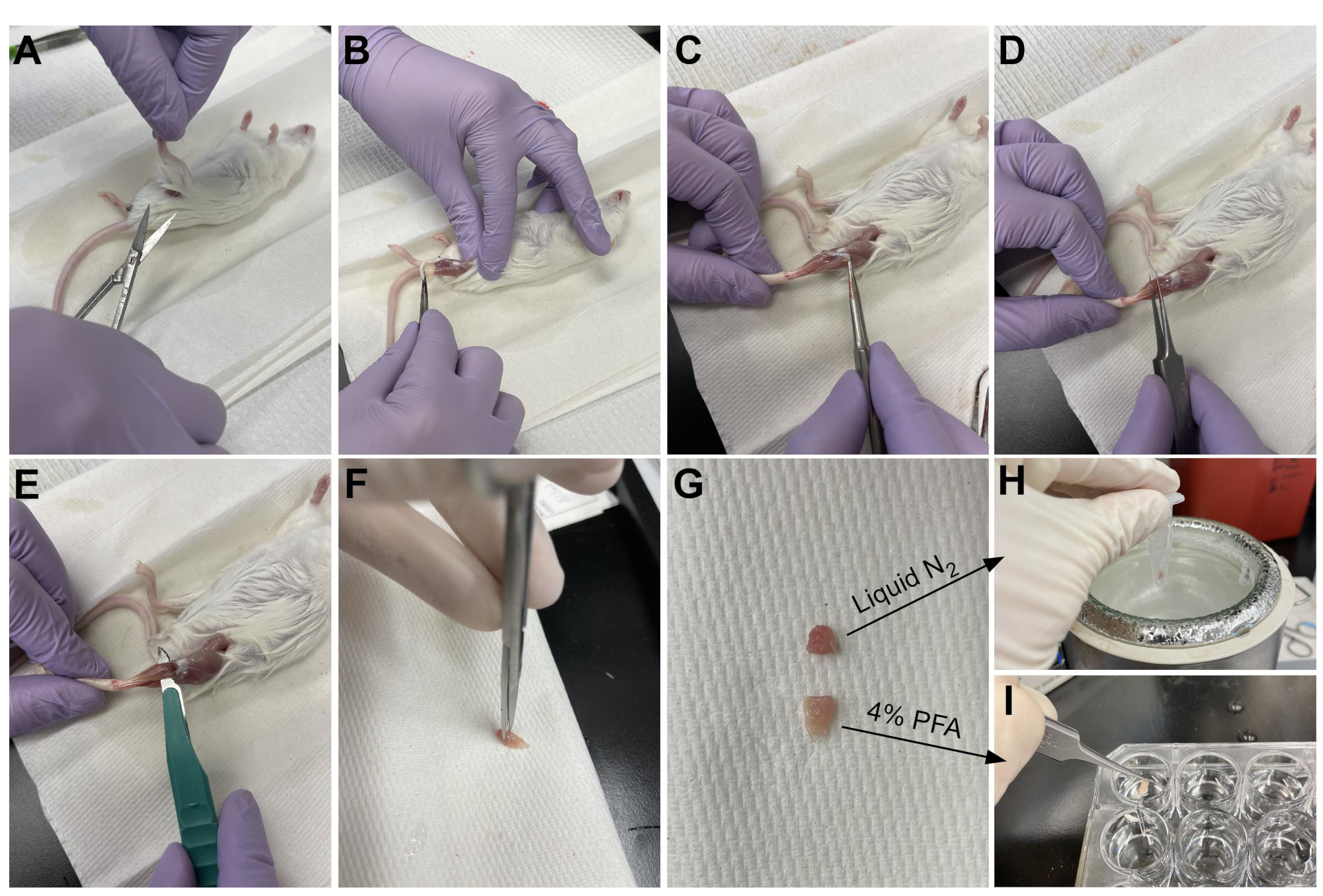
- Use scissors to cut the skin around the top of the leg, near the pelvis (Figure 3A).
- Gently pull the skin of the leg from the top down to the ankle (Figure 3B).
NOTE: The TA is a teardrop-shaped muscle with a clearly defined distal tendon attaching to the medial cuneiform and first metatarsal bone. It is lateral to the tibia and extends up to the lower knee. - First, remove the outer connective tissue layer (epimysium) using sharp-tipped tweezers prior to harvesting the TA (Figure 3C). Use a dissecting microscope to visualize the epimysium better.
- Slide the tweezers underneath the TA from the bottom of the muscle, starting at the distal tendon, and gently pull upward toward the knee (Figure 3D). Stop at the end of the muscle; do not push past the resistance felt at the lower knee.
- If there is significant resistance prior to reaching the lower knee, stop and continue removing leftover layers of connective tissue.
NOTE: There is another distal tendon just lateral to the TA tendon that attaches to the EDL, which is a slender muscle lateral to the TA. Being careful to only slide the tweezers under the TA tendon prevents accidental harvesting of the EDL, but it can also be easily removed after fixation. - Once the TA has been partially lifted from the leg with the tweezers, use the same motion with a scalpel to sever the connection of the TA to the lower knee (Figure 3E). Cut the tendon at the ankle with scissors to fully remove the TA. Only handle the muscle at the tendon to avoid damaging the fibers.
- Cut 1/3 of the TA at the end opposite the tendon (Figure 3F), put it in a microcentrifuge tube, and snap-freeze by dropping it into liquid nitrogen (Figure 3H).
- Submerge the other 2/3 of the tissue in a labeled well with 4% PFA for histology (Figure 3I). Be sure to keep track of when the first and last tissue was placed in fixative. Place on a shaker for 2-2.5 h at 4 °C.
- The duration of fixation is dependent on the tissue and its size. Determine the duration required for fixing the tissues. Fixing TAs for 2-2.5 h at 4 °C typically preserves adipocyte morphology well without causing over-fixation of the tissue.
NOTE: If planning to use TA for whole-mount immunofluorescent staining, skip the rest of this protocol until reaching Section 7: "Whole Mount Immunofluorescent Staining". - After fixation, remove PFA from the wells, rinse the tissues with cold 1x PBS 2-3 times, and then wash 2-3 times with cold 1x PBS for 5 min per wash.
- Remove the PBS from the wells and add enough 30% sucrose in 1x PBS to allow the tissue to float. Place on the shaker at 4 °C overnight.

Figure 3: Tissue harvest summary. (A) The skin is cut at the base of the leg and (B) the hind limb muscles are exposed. (C) Once the epimysium is removed from the TA, (D) forceps are used to partially separate the muscle and ensure the epimysium has been removed completely. (E) The TA is cut from the leg with a scalpel and removed after cutting the tendon. (G) After cutting the TA into a one-third and a two-thirds piece, (H) one-third is snap-frozen in liquid nitrogen for RT-qPCR analysis and (I) the other two-third is fixed in 4% PFA for histology. Please click here to view a larger version of this figure.
{kind=link}
4. Embedding
- Prepare the specimen molds to be used for embedding by labeling and filling with enough embedding medium to fully submerge the tissues.
- Remove tissues from the wells, dry off excess sucrose on a paper towel, and move to the freezing medium-filled specimen molds.
NOTE: It is helpful to know in what orientation the mold with be sectioned on the cryostat. This way, the user can orient the tissues in the mold in a way that allows the area of interest to be easily accessible. For the TA, this would be achieved by putting the thickest end (opposite from the tendon side) facing the surface that will be sectioned. This allows the TA to be easily sectioned at its thickest part (belly) and allows cross-sectional cutting of the muscle fibers. - Prepare an isopentane slurry by partially submerging a container holding isopentane into liquid nitrogen. Ensure there is enough liquid isopentane in the container to submerge about half of the specimen mold to be used to embed the tissues.
- Begin freezing the molds by carefully putting them in the isopentane slurry and making sure that about half of the mold is submerged. Also, make sure that the mold is freezing equally from all four sides.
- Take the mold out of the isopentane just before the entire mold is visibly frozen over from the top. The amount of time this takes depends on the molds being used.
- Keep the frozen molds in a container with dry ice while freezing the rest of the blocks, then store them at -80 °C.
NOTE: Isopentane for freezing can be reused. Put into a glass bottle but do not tighten the lid until isopentane reaches room temperature (RT). Otherwise, the change in pressure could shatter the bottle.
5. Sectioning
- Set the cryostat to -22 to -24 °C, add molds containing TAs into the cryostat, and wait for a minimum of 30 min for temperature acclimatization. In the meantime, label a series of positively charged microscope slides.
- Insert an anti-roll plate and align to the cryostat such that there are minimal nicks in the plate where it makes contact with the specimen block. Secure in place.
- Insert a fresh cryostat blade into the blade holder and secure it in place.
CAUTION: The blade is sharp. Cover the blade when manipulating other parts of the cryostat or frozen molds. - Remove the frozen block from the mold. Add a uniform layer of embedding medium to the cryostat chuck and position the block in the medium. Let it sit for 1-3 min until the embedding medium is completely frozen (opaque white).
NOTE: For TAs, the thickest area (belly) should be visible when on the chuck. - Place the cryostat chuck with the tissue block into the cryostat. Uncover the blade and advance the cryostat forward until just coming into contact with the blade. Section through the block at 25 µm sections until the tissue is no longer obscured by the embedding medium.
NOTE: While sectioning, adjust the angle of the cryostat and/or position of the stage such that the sections are of uniform thickness. It may be helpful to collect a few sections to ensure uniformity in section thickness. It is recommended that the user finds the proper anti-roll plate position prior to sectioning through to the region of interest within the TA (belly), as this allows the user to try several positions of the anti-roll plate until the sections are coming off straight without wasting tissue. - Change section thickness to 10-12 µm and collect the sections on labeled microscope slides. Serial sectioning is recommended by collecting adjacent sections on 6-10 slides (labeled 1-x), which allows for staining for multiple markers. If needed, use a thin brush to uncurl sections before collecting them on the slide.
NOTE: If sections are curling, check that the temperature is holding steady within the -22 to -24 °C range. If there are vertical striations in sections, this could be due to a nick in the anti-roll plate or the blade; this can be fixed by adjusting the position of the anti-roll plate and/or switching to a new blade. - After collecting adjacent sections of the same sectioning plane onto each slide, adjust the thickness back to 25 µm to advance 150-200 µm through the block, then adjust the thickness back to 10-12 µm and begin sectioning again.
NOTE: This serial sectioning allows the user to visualize, image, and quantify at different depths through the TA; three to four serial sections per slide is sufficient. - Store the slides and tissue blocks at -80 °C.
6. Immunofluorescent (IF) staining of tissue sections
NOTE: As antibody concentrations can vary between lots and manufacturers, optimization is recommended by evaluating several different concentrations of the antibodies on test slides prior to staining the slides of interest.
- Thaw/dry slides either at RT or on a warm plate at 37 °C for 10-20 min.
- Use a hydrophobic pen to draw a line at the edge of the slide's paper surface, where it meets the glass.
- Put the slides in a Coplin jar and wash with 1x PBS + 0.1% Tween20 (PBST) 3-5x on a shaker for at least 5 min per wash to rehydrate the tissue sections.
NOTE: At this point, it is important to not let the slides sit without being submerged in PBST (up to the hydrophobic line), or the tissue sections will dry out. - Place the slides on the rack of a humidifying chamber and overlay the slides with 310-350 µL of blocking solution (5% donkey serum and 0.3% Triton X-100 in 1x PBS) for 1-2 h at RT.
NOTE: No extra permeabilization step is needed, as the blocking solution contains 0.3% Triton X-100 allowing for sufficient permeabilization of the tissue sections. When using primary antibodies derived from mice (i.e., PAX7 and MYOD1 listed below), it is recommended to include a mouse-on-mouse blocking step using Fab fragments (1:50) in the blocking solution for the blocking step. This will help reduce background due to nonspecific binding of the mouse secondary antibody to antibodies other than the primary. - Dilute the primary antibodies to be used in the blocking solution shortly before proceeding to the next step as follows:
- Primary antibodies for adipocyte staining and whole section imaging: Dilute rabbit anti-Perilipin at a dilution ratio of 1:1000.
- Primary antibodies for lineage tracing of adipocytes: Dilute chicken anti-GFP at a ratio of 1:1000 and rabbit anti-Perilipin at 1:1000.
- Primary antibodies for lineage tracing of FAPs: Dilute chicken anti-GFP at a ratio of 1:1000 and goat anti-PDGFRα at 1:250.
- Primary antibodies for myogenic markers: Dilute mouse anti-PAX7 at a ratio of 1:25 or mouse anti-MYOD1 at 1:250, and rabbit anti-LAMININ at 1:1000.
NOTE: While the antibodies listed above have been successfully assessed with this protocol, it is likely that other markers and antibodies to label FAPs, adipocytes and/or other cell types are also compatible with this protocol. It is strongly recommended when using primary antibodies for the first time that the user include a negative control slide, in which primary antibodies are omitted. This will control for the specificity of the antibodies. All other steps in the protocol are followed, including the addition of secondary antibodies, but the blocking solution is used alone in the next step instead of the primary antibody in the blocking solution.
- Dump the blocking solution off the slides and overlay with 310-350 µL of blocking solution with primary antibodies and incubate overnight at 4 °C in the humidifying chamber.
- The next day, dump the blocking solution/primary antibodies off the slides and place them in a Coplin jar. Rinse the slides 2-3 times with PBST and wash 3-5x with PBST on a shaker for at least 5 min per wash.
- During the last wash, prepare the secondary antibodies or any direct conjugates to be used in the blocking solution. Minimize the amount of time these antibodies spend in the light to avoid photobleaching.
- Dilute secondary antibodies/direct conjugates: 488 nm donkey anti-chicken (1:1000) or anti-rabbit (1:1000) or anti-mouse (1:1000), 568 nm donkey anti-goat (1:1000) or anti-rabbit (1:1000) or Phalloidin (myofibers; 1:100), DAPI stain (nuclei; 1:500).
- Overlay the slides with 310-350 µL of the blocking solution with secondary antibodies and/or direct conjugates in the humidifying chamber. Incubate at RT for 1-2 h. Protect samples from light from now on.
- Dump the blocking solution/secondary antibodies and place them in a Coplin jar. Rinse with PBST once and wash 3-5x with PBST on a shaker for at least 5 min per wash. Keep the Coplin jar covered to prevent light exposure.
- Dry the slides as well as possible by tapping the edges and wiping the back against a paper towel, but do not let the tissue sections dry out.
- Add three or four drops of aqueous mounting medium to the top horizontal edge of the slide and gently add a coverslip. Do not press down or move if air bubbles form under the coverslip; any pressure or movement can distort the fragile cellular architecture of adipocytes.
- Allow mounting medium to set in the dark overnight before imaging.
7. Whole-mount immunofluorescent staining
- After ~1 h of fixation (see step 3.14), use sharp-tipped tweezers to peel away myofibers from the fixed TA.
- Place the separated fibers into a 24-well plate and wash 3x for 3 min each with PBST. For all subsequent incubations, ensure to add the lid to prevent evaporation.
- Incubate for 1 h in 1% Triton X-100 in 1x PBS at RT (200-300 µL) on a shaker to allow better penetration of antibodies.
- After rinsing a few times with PBST, overlay with blocking solution (200-300 µL) and block on a nutator or shaker overnight at 4 °C.
- Dilute the primary antibodies at the desired concentration (doubling the concentration tends to be a good starting point) in the blocking solution. Incubate the samples (200-300 µL) on a nutator or shaker overnight at 4 °C.
- Wash the samples rigorously with PBST throughout the day with frequent changes at RT on the shaker, about 4-6x for 30-60 min each wash.
- Dilute the secondary antibodies in the blocking solution at the desired concentration (1:500 tends to work well) plus nuclear staining and incubate the samples (200-300 µL) on a nutator or shaker overnight at 4 °C.
- Wash samples rigorously with PBST throughout the day with frequent changes at RT on the shaker, about 4-6x times for 30-60 min each wash or wash overnight at 4 °C.
- To mount, gently dry off excess PBST and then place the fibers in one or two drops of mounting medium on a glass slide (Figure 4A). To raise the coverslip (18 mm x 18 mm), add little clay feet; this will prevent the fibers from being squashed and secure the coverslip to the slide. Modeling compounds work well for this. Once the coverslip is secured, add more medium to the edge until the area under the coverslip is full.
NOTE: Instead of using a mounting medium containing anti-fading agents, the tissue can also be moved through an ascending series of glycerol (30% to 80% glycerol in PBS). - Wait for 1-2 days before imaging to allow curing of mounting medium.
8. Imaging of intramuscular fat
- Turn on the microscope and launch the imaging software. Secure the slide on the stage.
NOTE: For imaging adipocytes in muscle sections, a 5x or 10x objective combined with widefield microscopy is often sufficient. For visualizing WM-IF, a confocal microscope is required. - Use any channel to identify the area to be imaged.
- In the imaging software, adjust the gain and exposure time for each channel.
- Take images of the whole tissue in each channel (automatic or manual according to microscope and software used) and merge individual tiles to make a composite of the full TA cross-section.
NOTE: It is recommended to take images of two or three different sections of the same TA at different depths. By quantifying the adipocytes in each section and then reporting the average, localized differences in the amount of intramuscular fat due to, for example, injection errors will be avoided.
9. Quantifying adipocytes
- If not previously installed, add the Cell Counter plug-in to ImageJ (https://imagej.nih.gov/ij/plugins/cell-counter.html).
- Import the images into ImageJ as TIF files or original microscope files. View each channel in ImageJ as a separate TIF file.
NOTE: If using LIF or similar microscope file types, under Bio-Formats Import options, choose Hyperstack for View Stack With and check the box for Split Channels. Click OK to open the file. Also, ensure that the Autoscale box is unchecked. - Ensure that images for each channel are in 8-bit format (and gray): Image > Type > 8-bit.
- Merge DAPI (blue), GFP (green), PERILIPIN (red), and PHALLOIDIN (gray) images: Image > Color > Merge Channels.
- Check that scale (Analyze > Set Scale) is in microns. Using the freehand selection tool, outline the injured and uninjured portion of each cross-section, then measure (Analyze > Measure) and record the injured vs. uninjured area in a spreadsheet.
NOTE: Injured muscle can be identified as areas devoid of myofibers or areas populated by myofibers that contain centrally-located nuclei. - Launch Cell Counter: Plugins > CellCounter > Initialize.
- Select a counter type, then count each adipocyte. Record the total number of adipocytes in a spreadsheet, then calculate the number of adipocytes per 1 mm2 of the injured area.
10. Adipogenic gene expression analysis using RT-qPCR
- RNA Isolation
- Before beginning, preheat RNase-free water to 45 °C and prepare fresh 70% EtOH (350 µL per sample).
- Add 1,000 µL of guanidium thiocyanate to each tube containing the sample (see step 3.12.). It is important that bead beater-approved tubes are being used.
CAUTION: Guanidium thiocyanate is toxic. Wear appropriate personal protective equipment and handle in a fume hood. - Add three medium beads or one large and one small bead to each tube.
- Homogenize the tissue at 50 Hz for 2-4 min using a bead beater. Depending on tissue type and sample size, it may take up to 10 min.
- Add 200 µL of chloroform.
CAUTION: Chloroform is toxic. Wear personal protective equipment and handle in a fume hood. - Shake the samples for 15 s. Incubate for 2-3 min at RT.
- Centrifuge for 15 min at 12,000 x g. Pipette out 350 µL of the clear supernatant (upper layer containing the RNA) and add to a new microcentrifuge tube containing 350 µL of 70% ethanol. Be careful not to aspirate the lower protein and/or DNA layers
- Transfer up to 700 µL of the mixture to a mini spin column placed in a 2 mL collection tube. Continue with RNA isolation following the manufacturer's instructions.
- Elute with 30-50 µL of RNase-free water, depending on expected yield. Keep RNA on ice and measure the yield using a spectrophotometer. Keep the RNA stored at -80 °C
NOTE: It is possible to omit the DNase treatment step, as carefully taking off just the upper 350 µL of the RNA layer is sufficient in preventing DNA contamination. In addition to RT-qPCR analysis, the isolated RNA can also be used for RNA-sequencing, in which case a DNase treatment step is highly recommended.
- cDNA synthesis
- Use up to 1 µg of RNA to synthesize cDNA with a cDNA synthesis kit, following the manufacturer's instructions.
- After the run is complete, add 80 µL of RNase-free water. Store the samples at -20 °C.
- RT-qPCR of adipocyte-selective genes.
- Using a 384-well format, add 1 µL of primer (~1 µM final concentration) to the bottom of each well. Pre-drying primers results in tighter technical replicates. Leave covered until primers have evaporated completely (the plate can be placed on a heating block set to 37 °C to speed up evaporation).
- Set up sample reactions with four to eight technical replicates as follows: 2.5 µL of dye-based RT-PCR master mix, 2.1 µL of RNase-free water, and 0.4 µL of cDNA (~1 ng) with 5 µL total volume per well. The following thermal cycling conditions were used: denature at 95 °C for 15 s and anneal/extend at 60 °C for 25 s for 40 cycles.
- Normalize raw CT (cycle threshold) values to housekeeping genes (i.e., Hprt and Pde12) levels by calculating ΔΔCT as described here38. See20 for primer sequences.
NOTE: Standard practice for RT-qPCR analysis should be followed, such as the use of a minus Reverse Transcription (-RT) control, PCR reaction, and primer validation.
Results
Immunofluorescent visualization of intramuscular fat
Following the steps above and viewing Figure 1A, TA tissue sections were gathered from a 21 day post glycerol injury that were either snap-frozen immediately after harvesting in LN2-cooled isopentane or were fixed in 4% PFA for 2.5 h. After cryosectioning and staining both samples, images were taken at mid-belly, the largest area of the TA. PERILIPIN+ adipocytes from the unfixed TAs (Figure 1B) have significantly altered morphology compared to fixed sections (Figure 1C), making their identification, visualization, and subsequent quantification much more difficult and potentially inaccurate. To note, the first PERILIPIN+ lipid droplets were detected at around 5 days post-injury, with most adipocytes having formed by day 7. By 21 days post-injury, adipocytes had fully matured.
As the amount of fat per TA strongly correlates with the severity of the induced injury, the TAs must be injured significantly to effectively observe and study intramuscular fat formation. Practicing injections using ink into cadaver TAs is a great way to improve injury severity. Successful injuries tend to be above 50% of the muscle. To note, injured areas of the muscle represent areas devoid of muscle fibers or areas that are populated by muscle fibers that contain at least one centrally-located nucleus, a known hallmark of a regenerating muscle fiber.
This protocol can be readily adapted to stain for FAPs and fat in 3D. For this, multiple myofibers from the TA post-fixation were carefully separated, followed by whole-mount immunofluorescence. The key is to properly secure the fibers to the glass slide and, at the same time, to avoid over-compression of the tissue. By using moldable clay feet, the user can adjust the required thickness and secure the coverslip to the slide, even allowing the use of an inverted microscope (Figure 4A). This method was used successfully to label PDGFRα+ FAPs, Phalloidin+ myofibers, and PERILIPIN-expressing adipocytes (Figure 4B, Supplemental Video 1 and Supplemental Video 2). After obtaining images at multiple z-planes spanning up to 150 µm in thickness, the 3D rendering module within the microscope software was used to create a 3D reconstruction.

Figure 4: Whole-mount immunofluorescent staining. (A) Top and side view of how to mount the sample and add coverslip for whole-mount staining. (B) Representative 3D reconstructions of FAPs (green; left) and adipocytes (red; right) along with myofibers (gray) and nuclei (blue). Scale bars: 50 µm. Please click here to view a larger version of this figure.
{kind=link}
Quantification of intramuscular fat
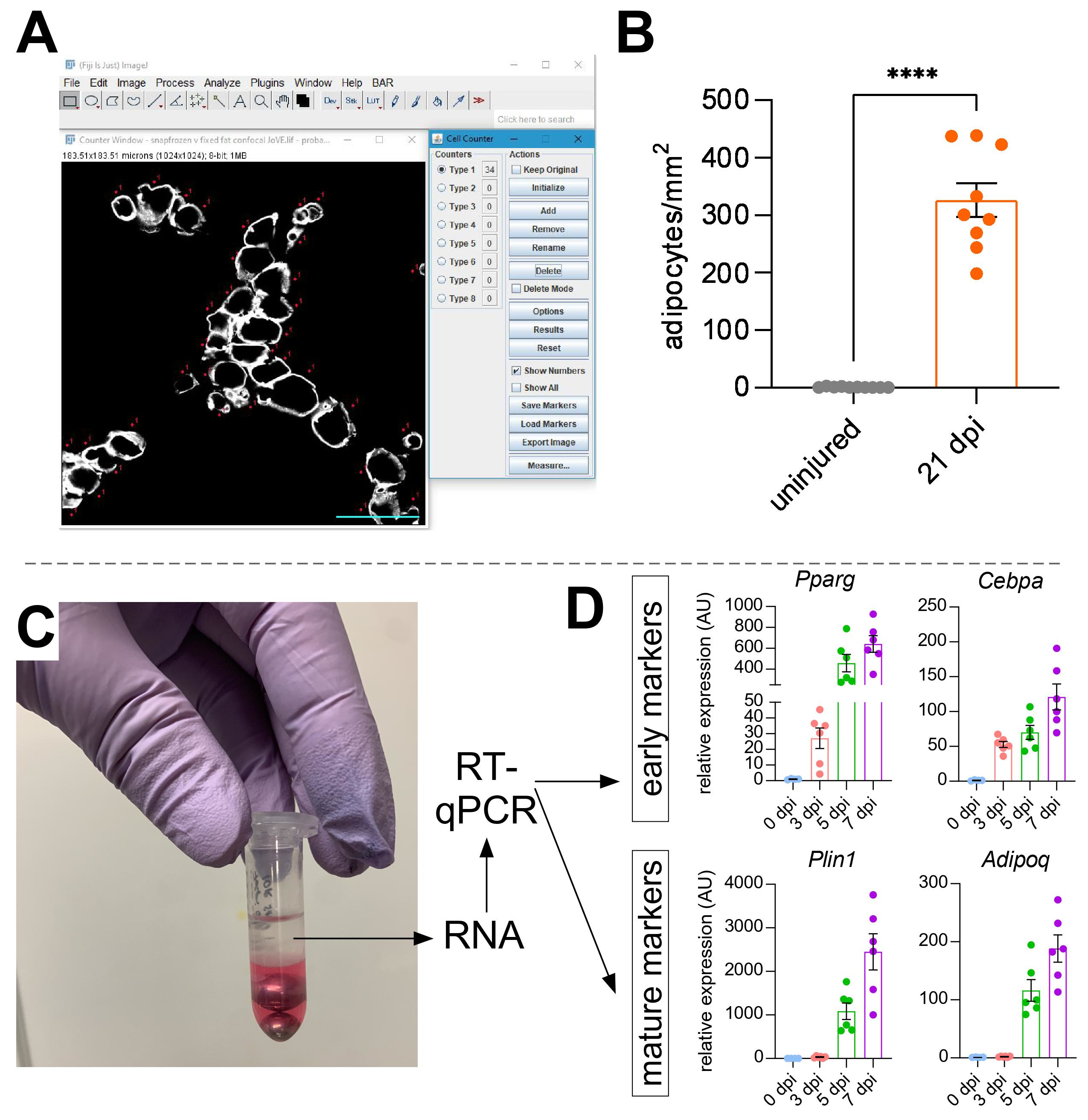
Once images have been taken of intramuscular fat, the Cell Counter function in ImageJ/FIJI was used to manually count the number of PERLIPIN+ adipocytes (Figure 5A). Next, the total area of the muscle section as well as the injured area, defined by centrally-located nuclei within myofibers, was determined. To control for injury severity, the total number of adipocytes was divided by the injured area resulting in the number of fat cells per 1 mm2 of injured muscle. Usually, TAs that display <30% injury are excluded from the quantifications. To note, although adipocytes are rare without injury, ranging from zero to eight per cross-sectional area, the total number of adipocytes are still normalized by total area. As highlighted in Figure 5B, a glycerol injury causes massive amounts of intramuscular fat compared to an uninjured TA muscle. Alternatively, as Perilipin staining is very clean with a high signal-to-noise ratio, it is also possible to use the Analyze Particle function to determine the total area occupied by Perilipin. However, this method will not be able to distinguish between smaller vs. fewer adipocytes. Up to three sections from a minimum of four individual animals were imaged and quantified, and the average number of fat cells present per mouse was reported.

Figure 5: Quantifications of intramuscular fat. (A) Representative image of how to count PERILIPIN+ adipocytes (white) using the Cell Counter function in ImageJ. Scale bar: 50 µm. (B) Whole TA adipocyte quantifications 21 days post glycerol injection normalized to 1 mm2 of the injured area. Each dot represents the average of one mouse. Error bars shown as SEM. **** = p < 0.0001. (C) RNA layer after homogenization and subsequent phase separation by chloroform is being used for RT-qPCR analysis. (D) Fold changes in expression levels of Pparg and Cepbα, early adipogenic genes, and Plin1 and Adipoq, two mature adipocyte markers, at different time points post glycerol injury. Each dot represents the average of one mouse. Error bars shown as SEM. Please click here to view a larger version of this figure.
{kind=link}
To independently confirm the amount of intramuscular fat present, gene expression levels of various adipogenic markers can be determined. For this, RNA can be isolated from a portion of the same TA muscle used for immunofluorescence (see steps above) at different points post-injury. A bead beater was used in combination with guanidium thiocyanate to homogenize the tissue. After adding chloroform followed by centrifugation, the upper RNA-containing layer was carefully extracted, and mini spin columns were used for RNA cleanup (Figure 5C). This method routinely produces high quality and quantity of RNA suitable for all downstream analyses such as RT-qPCR and RNAseq. For RT-qPCR, the relative expression levels of adipogenic to housekeeping genes were determined, and any relative changes were assessed following the ΔΔCT method38. As described in Figure 5D, compared to uninjured TA muscle, glycerol injury induces expression of early adipogenic markers such as Pparg and Cebpα as soon as 3 days post-injury. Mature markers, such as Adiponectin (Adipoq) and Perilipin (Plin1), can be detected as early as 5 days after glycerol injury.
Genetic lineage tracing of adipocytes
The adipocyte staining protocol presented here can be easily adapted to include genetic lineage tracing of FAPs to map and follow their fate into adipocytes. We have, for example, previously demonstrated that recombination could be induced via tamoxifen administration in PdgfrαCreERT2; Rosa26EYFP mice 2 weeks prior to the injury, effectively removing the floxed stop coding and indelibly activating EYFP expression in FAPs (Figure 6A). We achieved high recombination efficiencies with the tamoxifen regimen presented here, with ~75% of PDGFRα+ FAPs expressing EYFP20, similar to what other laboratories have reported27,39,40. Demonstrating that FAPs are indeed the cellular origin of intramuscular fat, the majority of FAPs have turned into EYFP+ PERILIPIN-expressing adipocytes 7 days post glycerol injury (Figure 6B).

Figure 6: Lineage tracing of FAPs. (A) Schematic overview of the experimental setup. (B) Representative immunofluorescent images showing successful recombination and activation of EYFP (yellow) within PDGFRα+ FAPs (red, arrowheads) and PERILIPIN+ adipocytes (red, asterisks). Scale bars: 25 µm. Please click here to view a larger version of this figure.
{kind=link}
Detection of multiple cell types
This protocol can also be used to visualize the myogenic compartment. Using antibodies against PAX7 and MYOD1, muscle stem cells (MuSCs) and myoblasts, respectively, can be readily detected 5 days post glycerol injury even in PFA-fixed muscle tissue section (Figure 7). Thus, the presented protocol is versatile and adaptable to not only label and image adipocytes and FAPs but also other cell types of the myogenic lineage.

Figure 7: Muscle stem cell and myoblast immunofluorescent staining. (A) Schematic overview of the experimental setup. (B) Representative immunofluorescent images showing successful staining of muscle stem cell (MuSC) (yellow, left) with PAX7 and myoblasts (yellow, right) with MYOD1. LAMININ outlines the myofibers (white), and nuclei are in cyan. Scale bars: 50 µm. Please click here to view a larger version of this figure.
{kind=link}
Supplemental Video 1: 3D rendering of FAPs. Three-dimensional reconstruction of myofibers, FAPs, and nuclei stained for PHALLOIDIN (gray), PDGFRα (green), and DAPI (blue), respectively, 21 days post injury. Please click here to download this Video.
Supplemental Video 2: 3D rendering of intramuscular fat. Volumetric rendering of myofiber bundles (gray, PHALLOIDIN) and intramuscular fat (red, PERILIPIN), which has replaced a myofiber 21 days post glycerol injury. Please click here to download this Video.
Discussion
This protocol outlines an extensive and detailed protocol that allows for efficient visualization and rigorous quantification of intramuscular fat. By splitting the same muscle into two parts, one being used for immunofluorescence and the other for RT-qPCR analysis, this protocol is also very versatile. It can also be combined with genetic lineage tracing of FAPs to study their conversion into adipocytes under certain conditions and is highly adaptable to label and image multiple additional cell types.
The most commonly used ways to visualize intramuscular fat are paraffin sections followed by hematoxylin and eosin staining or frozen sections stained for lipophilic dyes such as Oil Red O (ORO). However, while paraffin-processed tissues maintain the best histology, the same process also extracts all lipids preventing the use of lipophilic dyes. Although lipophilic staining methods will work on both PFA-fixed and unfixed tissue sections, lipid droplets are easily displaced by applying pressure to the coverslip, thereby distorting the spatial distribution of intramuscular fat. To circumvent this, a recent study established a rigorous protocol to visualize ORO+ adipocytes using a whole-mount approach. For this, the authors decellularized the TA to visualize the spatial distribution of intramuscular fat throughout the whole TA41. As powerful as this technique is, it also prevents the use of other co-stains to mark additional cellular structures. The whole mount immunofluorescence approach presented here can be used to co-stain adipocytes with a variety of markers allowing for fine mapping of the cellular environment. One major challenge, however, is tissue penetration of the antibodies. The more fibers are kept together, the more difficult it will be for the antibodies to equally penetrate and bind all available antigens. Thus, this method is most effective when looking at small groups of fibers. At the same time, this is also a limitation as the overall anatomical location of intramuscular fat is being lost when focusing on only small, peeled-off fiber bundles. However, with the current development of novel tissue clearing methods plus new imaging technology, greater tissue penetration and visualization will be possible in the future42,43,44.
While prior fixation of muscle tissue preserves adipocyte morphology, it also creates a challenge to assess the size of myofibers, an important measurement of muscle health. Myofiber size is determined by measuring the cross-sectional area of myofibers. We have previously reported that prior fixation of muscle tissue will cause most markers available to outline myofibers to fail31. To overcome this hurdle, we have developed a novel image segmentation pipeline, which allows the measurement of myofiber size even in fixed muscle sections31. Thus, we have established a robust and efficient tissue processing pipeline that, combined with this protocol, overcomes most disadvantages caused by prior fixation of muscle tissue.
Another major advantage of this approach is versatility. By splitting the TA into two parts, the amount of information that can be obtained from one muscle is maximized. This not only reduces animal numbers but also adds an extra layer of control by confirming histology through gene expression and vice versa. In addition, many different genes can be examined beyond adipogenic genes. The isolated RNA can also be used for a whole muscle RNAseq experiment. Finally, the snap-frozen muscle piece can also be used for protein work. One limitation of this protocol is the possibility of the injury not being consistent across the full length of the TA. This could lead to a scenario where the two muscle parts diverge in the amount of intramuscular fat they contain and may warrant exclusion of such a sample from any downstream analysis. It is, therefore, recommended to not simply rely on RT-qPCR to draw major conclusions about the amount of intramuscular fat, but rather as supportive data to the histological quantifications.
Together, this protocol outlines a robust, efficient, and rigorous tissue processing pipeline that will allow visualization and quantification of intramuscular fat, the first step in developing novel treatment options to combat fatty fibrosis. At the same time, it is versatile and can be adapted to many different cell types within the muscle as well as adipocytes in other tissues.
Disclosures
The authors declare no competing interests.
Acknowledgements
We thank the members of the Kopinke laboratory for helping with data collections and critical reading of the manuscript. We also thank the members of the Myology Institute at the University of Florida for their valuable input on the manuscript. The work was supported by the NIH grant 1R01AR079449. Figure 2 was created with Biorender.
Materials
| Name | Company | Catalog Number | Comments |
| 16% PFA (Pack of 12, 10 mL bottles) | Electron Miscroscopy Sciences | 15710 | |
| 2.0 mL Microcentrifuge Tubes | Fisher Scientific | 05-408-138 | microcentrifuge tubes for snapfreezing/bead beating |
| 2-Methylbutane (4 L) | Fisher Chemical | O3551-4 | isopentane |
| Absolute Ethanol (200 proof) | ThermoFisher Scientific | BP2818100 | |
| AffiniPure Fab fragment donkey anti-mouse | Jackson ImmunoResearch | 715-007-003 | mouse-on-mouse blocking |
| Alexa Fluor 488 donkey anti-chicken secondary antibody | Jackson ImmunoResearch | 703-545-155 | |
| Alexa Fluor 488 donkey anti-mouse secondary antibody | Invitrogen | A21202 | |
| Alexa Fluor 488 donkey anti-rabbit secondary antibody | Invitrogen | A21206 | |
| Alexa Fluor 568 donkey anti-goat secondary antibody | Invitrogen | A11057 | |
| Alexa Fluor 568 donkey anti-rabbit secondary antibody | Invitrogen | A11037 | |
| Alexa Fluor 568 Phalloidin antibody | Invitrogen | A12380 | Dissolved in 1.5 mL methanol (~66 µM working solution) |
| BioLite 24-well Multidishes | ThermoFisher Scientific | 930186 | 24 well plate for PFA tissue incubation |
| Biometra TOne | analytikjena | 8462070301 | Thermal cycler |
| Chicken anti-GFP antibody | Aves Labs | GFP-1020 | |
| Chloroform/isoamyl alcohol 24:1(v/v) for molecular biology, DNAse, RNAse, and Protease free | ThermoFisher Scientific | AC327155000 | |
| Corn oil | Sigma Aldrich | C8267 | |
| DAPI stain | Invitrogen | D1306 | 150 µM working solution in dH2O |
| Donkey Serum (100 mL) | Millipore Sigma | 5058837 | for blocking solution |
| Dumont #5 Forceps | Fine Science Tools | 11251-20 | sharp-tipped tweezers |
| Fine Scissors Straight 9 cm | Fine Science Tools | 14060-09 | |
| Fluoromount-G | SouthernBiotech | 0100-01 | mounting medium |
| Glycerol, 99.5%, for molecular biology (500 mL) | Acros Organics | 327255000 | |
| Goat anti-PDGFRα antibody | R&D | AF1062 | |
| Hybridization Oven | VWR | 230301V | for Tamoxifen incubation |
| ImmEdge Hydrophobic Barrier PAP Pen | Vector Laboratories | H-4000 | hydrophobic pen |
| Insulin Syringe with Micro-Fine IV needle (28 G) | BD | 329461 | |
| Insulin Syringe with Slip Tip, 1 mL | BD | 329654 | Insulin syringe without needle, for oral gavaging |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 | |
| Isoflurane | Patterson Veterinary | 78938441 | |
| Leica DMi8 inverted microscope | Leica | micrscope used for widefield IF and confocal imaging | |
| Micro Slides | VWR | 48311-703 | positively charged microscope slides |
| mouse anti-MYOD antibody | Invitrogen | MA1-41017 | |
| mouse anti-PAX7 antibody (supernatant) | DSHB | AB 428528 | |
| MX35 Premier+ Microtome blades | ThermoFisher Scientific | 3052835 | microtome blades |
| NanoDrop 2000 Spectrophotometer | ThermoFisher Scientific | ND2000 | spectrophotometer for RNA yield |
| Play-Doh | Hasbro | modeling compound | |
| PowerUp SYBR Green Master Mix | ThermoFisher Scientific | A25742 | green dye PCR master mix |
| Puralube Vet Ointment | Puralube | 17033-211-38 | vet ophthalmic ointment |
| QuantStudi 6 Flex Real-Time 384-well PCR System | Applied Biosystems | 4485694 | qPCR machine |
| Rabbit anti-perilipin antibody | Cell Signaling Technology | 9349S | |
| Red-Rotor Shaker | Hoefer Scientific | PR70-115V | shaker for IF staining |
| Richard-Allan Scientific Slip-Rite Cover Glass | ThermoFisher Scientific | 152460 | coverslips |
| RNeasy Mini Kit | QIAGEN | 74106 | contains mini spin columns |
| Safe-Lock Tubes 1.5 ml, natural | Eppendorf | 22363204 | |
| Sample Tubes RB (2 mL) | QIAGEN | 990381 | |
| Sodium azide | Alfa Aesar | 14314 | |
| Stainless Steel Beads, 2.8 mm | Precellys | KT03961-1-101.BK | small beads |
| Stainless Steel Beads, 5 mm | QIAGEN | 69989 | medium beads |
| Stainless Steel Beads, 7 mm | QIAGEN | 69990 | large beads |
| Stainless Steel Disposable Scalpels | Miltex | 327-4102 | scalpel |
| Stainless steel feeding tube, 20 G x 38 mm, straight | Instech Laboratories | FTSS-20S-3 | gavage needle |
| Tamoxifen | Toronto Research Chemicals | T006000 | |
| Tissue Plus O.C.T. Compound | Fisher HealthCare | 4585 | embedding medium |
| TissueLyser LT | QIAGEN | 85600 | bead beater |
| TissueLyser LT Adapter, 12-Tube | QIAGEN | 69980 | |
| Tissue-Tek Cryomold | Sakura | 4566 | specimen molds |
| Triton X-100 | Alfa Aesar | A16046 | |
| TRIzol Reagent | ThermoFisher Scientific | 15596026 | guanidium thiocyanate |
| Tween20 (500 mL) | Fisher BioReagents | BP337-500 | |
| VWR Micro Slides - Superfrost Plus | VWR | 48311703 | |
| Wheaton Coplin staining jars | Millipore Sigma | S6016 | Coplin jar |
References
- Milad, N., et al. Increased plasma lipid levels exacerbate muscle pathology in the mdx mouse model of Duchenne muscular dystrophy. Skeletal Muscle. 7 (1), 19 (2017).
- Goodpaster, B. H., et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Archives of Internal Medicine. 165 (7), 777-783 (2005).
- Goodpaster, B. H., et al. Association between regional adipose tissue distribution and both type 2 diabetes and impaired glucose tolerance in elderly men and women. Diabetes Care. 26 (2), 372-379 (2003).
- Goodpaster, B. H., et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences. 61 (10), 1059-1064 (2006).
- Goodpaster, B. H., Thaete, F. L., Kelley, D. E. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. American Journal of Clinical Nutrition. 71 (4), 885-892 (2000).
- Goodpaster, B. H., Theriault, R., Watkins, S. C., Kelley, D. E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism. 49 (4), 467-472 (2000).
- Burakiewicz, J., et al. Quantifying fat replacement of muscle by quantitative MRI in muscular dystrophy. Journal of Neurology. 264 (10), 2053-2067 (2017).
- Murphy, W. A., Totty, W. G., Carroll, J. E. MRI of normal and pathologic skeletal muscle. American Journal of Roentgenology. 146 (3), 565-574 (1986).
- Willcocks, R. J., et al. Multicenter prospective longitudinal study of magnetic resonance biomarkers in a large duchenne muscular dystrophy cohort. Annals of Neurology. 79 (4), 535-547 (2016).
- Wokke, B. H., et al. Quantitative MRI and strength measurements in the assessment of muscle quality in Duchenne muscular dystrophy. Neuromuscular Disorders. 24 (5), 409-416 (2014).
- Contreras, O., Rossi, F. M. V., Theret, M. Origins, potency, and heterogeneity of skeletal muscle fibro-adipogenic progenitors-time for new definitions. Skeletal Muscle. 11 (1), 16 (2021).
- El Agha, E., et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 21 (2), 166-177 (2017).
- Joe, A. W., et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature Cell Biology. 12 (2), 153-163 (2010).
- Liu, W., Liu, Y., Lai, X., Kuang, S. Intramuscular adipose is derived from a non-Pax3 lineage and required for efficient regeneration of skeletal muscles. Developmental Biology. 361 (1), 27-38 (2012).
- Scott, R. W., Arostegui, M., Schweitzer, R., Rossi, F. M. V., Underhill, T. M. Hic1 defines quiescent mesenchymal progenitor subpopulations with distinct functions and fates in skeletal muscle regeneration. Cell Stem Cell. 25 (6), 797-813 (2019).
- Uezumi, A., et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. Journal of Cell Science. 124, 3654-3664 (2011).
- Hogarth, M. W., et al. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nature Communications. 10 (1), 2430 (2019).
- Uezumi, A., Fukada, S., Yamamoto, N., Takeda, S., Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nature Cell Biology. 12 (2), 143-152 (2010).
- Stumm, J., et al. Odd skipped-related 1 (Osr1) identifies muscle-interstitial fibro-adipogenic progenitors (FAPs) activated by acute injury. Stem Cell Research. 32, 8-16 (2018).
- Kopinke, D., Roberson, E. C., Reiter, J. F. Ciliary Hedgehog signaling restricts injury-induced adipogenesis. Cell. 170 (2), 340-351 (2017).
- Huang, Y., Das, A. K., Yang, Q. Y., Zhu, M. J., Du, M. Zfp423 promotes adipogenic differentiation of bovine stromal vascular cells. PLoS One. 7 (10), 47496 (2012).
- Sun, Y. -. M., et al. PDGFRα regulated by miR-34a and FoxO1 promotes adipogenesis in porcine intramuscular preadipocytes through Erk signaling pathway. International Journal of Molecular Sciences. 18 (11), 2424 (2017).
- Te, L. J. I., Doherty, C., Correa, J., Batt, J. Identification, isolation, and characterization of fibro-adipogenic progenitors (FAPs) and myogenic progenitors (MPs) in skeletal muscle in the rat. Journal of Visualized Experiments: JoVE. (172), e61750 (2021).
- Lukjanenko, L., et al. Aging disrupts muscle stem cell function by impairing matricellular WISP1 secretion from fibro-adipogenic progenitors. Cell Stem Cell. 24 (3), 433-446 (2019).
- Santini, M. P., et al. Tissue-resident PDGFRalpha(+) progenitor cells contribute to fibrosis versus healing in a context- and spatiotemporally dependent manner. Cell Reports. 30 (2), 555-570 (2020).
- Uezumi, A., et al. Identification and characterization of PDGFRalpha+ mesenchymal progenitors in human skeletal muscle. Cell Death & Disease. 5, 1186 (2014).
- Wosczyna, M. N., et al. Mesenchymal stromal cells are required for regeneration and homeostatic maintenance of skeletal muscle. Cell Reports. 27 (7), 2029-2035 (2019).
- Soliman, H., et al. Pathogenic potential of Hic1-expressing cardiac stromal progenitors. Cell Stem Cell. 26 (2), 205-220 (2020).
- Chung, M. I., Bujnis, M., Barkauskas, C. E., Kobayashi, Y., Hogan, B. L. M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development. 145 (9), (2018).
- Hilgendorf, K. I., et al. Omega-3 fatty acids activate ciliary FFAR4 to control adipogenesis. Cell. 179 (6), 1289-1305 (2019).
- Waisman, A., Norris, A. M., Elías Costa, M., Kopinke, D. Automatic and unbiased segmentation and quantification of myofibers in skeletal muscle. Scientific Reports. 11 (1), 11793 (2021).
- Kang, S. H., Fukaya, M., Yang, J. K., Rothstein, J. D., Bergles, D. E. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 68 (4), 668-681 (2010).
- Srinivas, S., et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology. 1, 4 (2001).
- Lukjanenko, L., Brachat, S., Pierrel, E., Lach-Trifilieff, E., Feige, J. N. Genomic profiling reveals that transient adipogenic activation is a hallmark of mouse models of skeletal muscle regeneration. PLoS One. 8 (8), 71084 (2013).
- Mahdy, M. A., Lei, H. Y., Wakamatsu, J., Hosaka, Y. Z., Nishimura, T. Comparative study of muscle regeneration following cardiotoxin and glycerol injury. Annals of Anatomy = Anatomischer Anzeiger: Official Organ of the Anatomische Gesellscaft. 202, 18-27 (2015).
- Pisani, D. F., Bottema, C. D., Butori, C., Dani, C., Dechesne, C. A. Mouse model of skeletal muscle adiposity: a glycerol treatment approach. Biochemical and Biophysical Research Communications. 396 (3), 767-773 (2010).
- Kawai, H., et al. Experimental glycerol myopathy: a histological study. Acta Neuropathologica. 80 (2), 192-197 (1990).
- Schmittgen, T. D., Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nature Protocols. 3 (6), 1101-1108 (2008).
- Uezumi, A., et al. Mesenchymal Bmp3b expression maintains skeletal muscle integrity and decreases in age-related sarcopenia. The Journal of Clinical Investigation. 131 (1), 139617 (2021).
- Biferali, B., et al. Prdm16-mediated H3K9 methylation controls fibro-adipogenic progenitors identity during skeletal muscle repair. Science Advances. 7 (23), 9371 (2021).
- Biltz, N. K., Meyer, G. A. A novel method for the quantification of fatty infiltration in skeletal muscle. Skeletal Muscle. 7 (1), (2017).
- Vieites-Prado, A., Renier, N. Tissue clearing and 3D imaging in developmental biology. Development. 148 (18), (2021).
- Gómez-Gaviro, M. V., Sanderson, D., Ripoll, J., Desco, M. Biomedical applications of tissue clearing and three-dimensional imaging in health and disease. iScience. 23 (8), 101432 (2020).
- Ueda, H. R., et al. Tissue clearing and its applications in neuroscience. Nature Reviews Neuroscience. 21 (2), 61-79 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved