
Method Article
Lineage Tracing and Clonal Analysis in Developing Cerebral Cortex Using Mosaic Analysis with Double Markers (MADM)
In This Article
Summary
A protocol to perform lineage tracing and functional genetic analysis of candidate genes at a single cell level using mosaic analysis with double markers (MADM) is presented. MADM clonal analysis provides a quantitative framework to measure the proliferative behavior, cellular output, and lineage relationship of individual progenitors and their daughter cells.
Abstract
Beginning from a limited pool of progenitors, the mammalian cerebral cortex forms highly organized functional neural circuits. However, the underlying cellular and molecular mechanisms regulating lineage transitions of neural stem cells (NSCs) and eventual production of neurons and glia in the developing neuroepithelium remains unclear. Methods to trace NSC division patterns and map the lineage of clonally related cells have advanced dramatically. However, many contemporary lineage tracing techniques suffer from the lack of cellular resolution of progeny cell fate, which is essential for deciphering progenitor cell division patterns. Presented is a protocol using mosaic analysis with double markers (MADM) to perform in vivo clonal analysis. MADM concomitantly manipulates individual progenitor cells and visualizes precise division patterns and lineage progression at unprecedented single cell resolution. MADM-based interchromosomal recombination events during the G2-X phase of mitosis, together with temporally inducible CreERT2, provide exact information on the birth dates of clones and their division patterns. Thus, MADM lineage tracing provides unprecedented qualitative and quantitative optical readouts of the proliferation mode of stem cell progenitors at the single cell level. MADM also allows for examination of the mechanisms and functional requirements of candidate genes in NSC lineage progression. This method is unique in that comparative analysis of control and mutant subclones can be performed in the same tissue environment in vivo. Here, the protocol is described in detail, and experimental paradigms to employ MADM for clonal analysis and lineage tracing in the developing cerebral cortex are demonstrated. Importantly, this protocol can be adapted to perform MADM clonal analysis in any murine stem cell niche, as long as the CreERT2 driver is present.
Introduction
The cerebral cortex is a highly organized structure composed of six distinct layers. The cortex contains a diverse array of cell types including neurons and glia, which interact to form functional neural circuits. Most, if not all, cortical excitatory projection neurons and glia are derived from a common pool of neural stem cells (NSCs) known as the radial glial progenitors (RGPs)1,2,3. RGPs themselves are derived from neuroepithelial stem cells (NESCs) composing the early embryonic neuroepithelium. By embryonic day 9 (E9) in mice, NESCs begin to transition into RGPs4. RGP lineage progression requires precise temporal and spatial regulation, and when this process is hindered, severe neurological disorders such as megalencephaly, microcephaly, lissencephaly, or impairments such as schizophrenia and autism can result5,6. At E10, most RGPs undergo symmetric proliferative divisions, resulting in an expansion of the neural progenitor pool4,7. RGPs eventually begin to divide asymmetrically, producing cortical projection neurons in a temporally defined manner. Through consecutive waves of neurogenesis, newborn neurons migrate into the cortical plate forming cortical laminae with early born neurons occupying deep layers and late born neurons residing in the superficial layers8,9,10. Because clonally related pyramidal neurons migrate radially into the cortex with very little tangential dispersion, daughter cells tend to form a column or cone shaped structure referred to as a neuronal radial unit4,11,12,13. By E17, embryonic neurogenic expansion is complete in mice14. RGPs can also produce ependymal cells and some classes of glia, including astrocytes and oligodendrocytes1,15,16,17,18,19. The potential of RGPs to give rise to both neurons and astrocytes seems to be consistent across all cortical regions18, with approximately 1/6 of neurogenic RGPs also producing glia11.
Currently, the genetic and epigenetic factors regulating temporal progression of a stem cell along its lineage are mostly unknown. Temporal patterns of gene expression may have substantial impact on lineage decisions in RGPs20,21,22,23,24. How this tightly knit relationship between temporal and spatial patterning leads to the molecular diversity of adult neuronal types across cortical areas is not known. Likewise, how the individual stem cell potential and its cellular output is modulated at the cellular and molecular level is an important unanswered question. Future studies will hopefully address some of these questions, ultimately expanding our understanding of functional cortical circuit formation.
Developmental neurobiology seeks to understand the lineage relationship that cells in the brain share with one another. Initially, very few research tools were available for this, and many early studies relied on visual observations of division patterns in transparent organisms such as Caenorhabditis elegans25. Recent decades have seen a dramatic increase in the number and sophistication of techniques available13,26,27,28,29. The emergence of the CRISPR-Cas9 genome editing system allows for synthetic reconstruction of cell lineage relationships by introducing evolving DNA barcodes27,30. Two recent examples of barcoding strategies include the use of homing guide RNA that directs CRISPR-Cas9 to specific DNA barcode loci or a cytidine deaminase fused with nickase Cas9 to target endogenous interspersed repeat regions31,32. These technologies provide highly multiplexed approaches through the introduction of barcodes that progressively and stably accumulate unique mutations over time. Genome editing approaches are highly valuable because they allow retroactive analysis of the relationship between any two cells based on the shared inheritance of these barcodes. However, in order to read the barcodes in individual cells, the tissue usually must be disrupted, and therefore information regarding position, morphology, and absolute cell numbers from an individual progenitor is lost.
Combinatorial labeling paradigms preserve spatial information and in principle also allow for the distinction between closely localized or even overlapping clones33,34. For a lineage tracing method to be informative it must label individual progenitors and their progeny in a sparse and indelible manner. Notably, the Brainbow35 and Confetti36,37 approaches use stochastic multicolor Cre recombinase-based reporters that express a combination of fluorescent proteins from a single locus. The extensive number of simultaneous color combinations that can be achieved in vivo make this a powerful tool when tracing cortical RGP clones and astrocytes34. Transposon-based systems providing stable genomic integration of transgenes encoding fluorescent reporters and permitting lineage tracing of cortical progenitors have also been developed33,38,39,40,41. Transposon-based systems have an added advantage in that the reporter constructs stably integrate into the genome, and thereby reliably label lineally related daughter cells. To trace astrocyte lineages specifically, a number of methods have been developed that involve electroporation of piggyBac transposases including Star Track, which makes use of a combination of constructs encoding different fluorescent proteins40,42. Another approach, MAGIC markers, introduces Brainbow vectors as transposable transgenes. This has been successfully used to trace embryonic neural and astrocyte progenitors34,43. Recently, mosaic analysis by dual recombinase-mediated cassette exchange (MADR) was found to stably label mutant cells expressing transgenic elements from precisely defined chromosomal loci44. These powerful in vivo combinatorial labeling techniques have provided numerous insights into the lineage dynamics of progenitor cells. However, these analyses are performed on fixed tissue, providing a snapshot of individual clones at a defined developmental stage. In order to observe changes in the lineage dynamics of single clones over time, chronic in vivo imaging methods similar to those performed in the adult dentate gyrus need to be applied45.
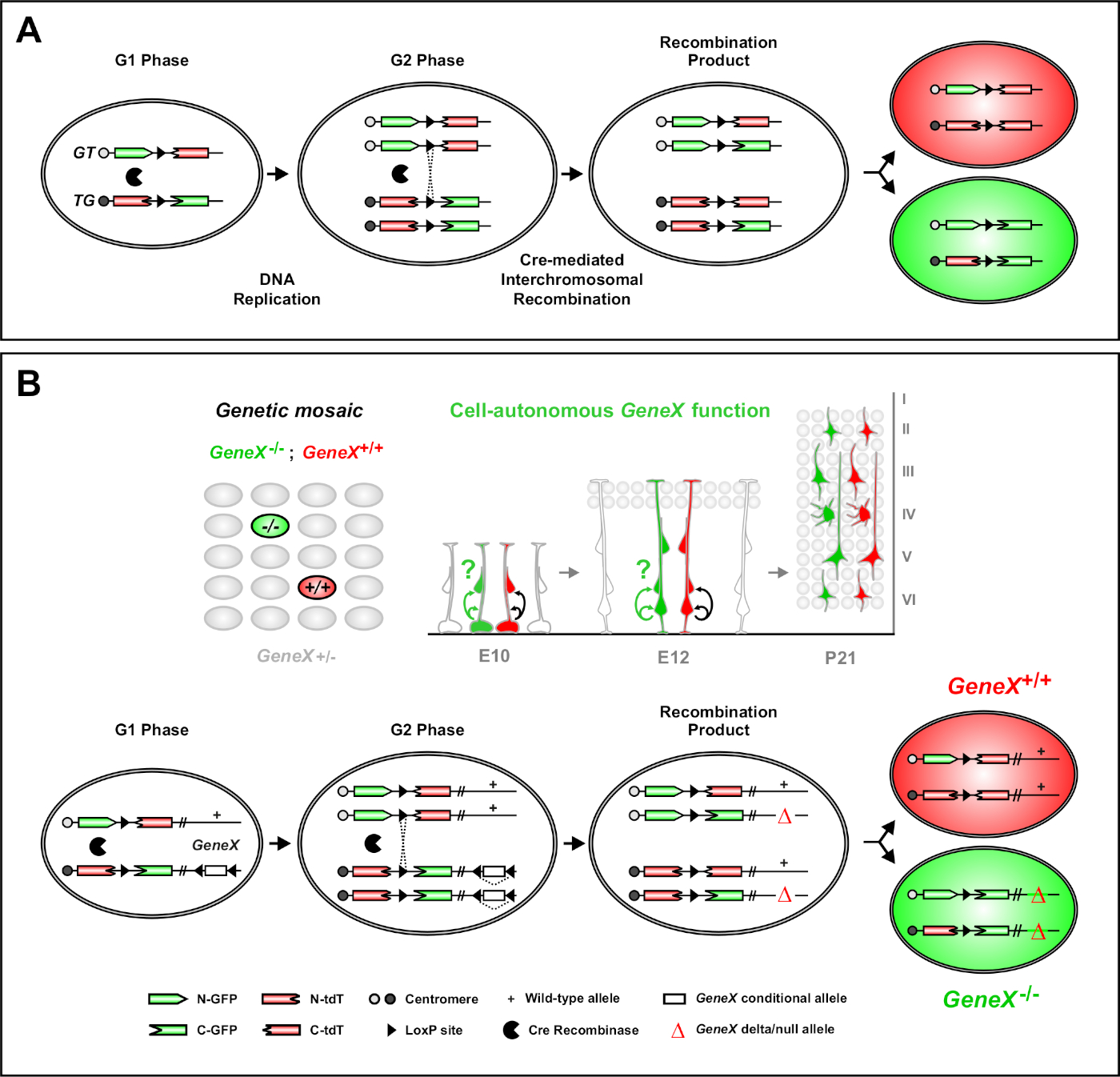
Mosaic analysis with double markers (MADM) is a powerful dual color labeling method that enables in vivo lineage tracing of individual progenitor cells in mice46,47. Two components are necessary for MADM labeling events to occur: First, MADM cassettes must be targeted to identical loci on homologous chromosomes. Cassettes consist of two chimeric fluorescent reporter genes, eGFP (green, [G]) and tandem dimer Tomato (red, tdT[T]). The GT cassette contains the N-terminus of eGFP and the C-terminus of tdT, separated by an intron containing a loxP site. The TG cassette is constructed inversely, with the N-terminus of tdT and the C-terminus of eGFP. Second, the expression of Cre recombinase in the same cell containing the targeted MADM cassettes is essential. In the absence of Cre, the chimeric cassettes do not express functional eGFP or tdT because their coding sequences are disrupted. The loxP sites serve as the target for Cre-mediated interchromosomal recombination, resulting in the reconstitution of both expression cassettes simultaneously. If recombination occurs during the G2 phase of cell cycle followed by X segregation (G2-X), the two daughter cells will each express one of the two fluorescent proteins. Temporal regulation of CreERT2 activity using tamoxifen (TM) provides precise information on birth date of MADM clones and the division patterns of their progeny (Figure 1A)29,46,47.
MADM can potentially systematically label individual clones with high single cell resolution in the mouse brain similar to traditional but nonspecific and laborious methods like Golgi staining48 or dye filling49. Because only the promoter driving CreERT2 determines the cell type specificity of clonal MADM labeling, MADM can in principle be applied for clonal lineage tracing throughout any murine organ and tissue47,50,51,52. Indeed, studies have already used MADM to reveal lineage relationships in clones derived from diverse tissues47,50,51,52,53,54,55,56,57,58,59. MADM experimental paradigms have been applied to study lineage in cortical projection neurons, glia, and postnatal stem cells in the developing neocortex7,11,12,46,60,61,62,63,64,65. MADM has also been used to study cell lineage in the adult dentate gyrus, thalamus, cerebellar granule cells, and interneurons at the clonal level (see Table 1 for a complete list)47,53,54,56,57,66.
A unique feature of MADM is the ability to genetically link mutations distal to one MADM cassette, thereby creating a genetic mosaic (Figure 1B and Figure 2). This results in wild type daughter cells labeled with one fluorescent marker (tdT in Figure 1B) and homozygous mutant siblings with the other (eGFP in Figure 1B) in an unlabeled heterozygous environment. MADM is unique in that comparative analysis of control and mutant subclones can be performed in the same tissue environment in vivo. Originally, MADM cassettes were targeted into the Rosa26 locus47, but MADM analysis of gene function was limited to genes distal to the locus. To overcome (at least partly) this limitation and expand the possibilities for MADM-based gene analyses, MADM cassettes were knocked in close to the centromeres of Chr. 751, Chr. 1146, and Chr. 1251. Targeting all 19 mouse autosomes with MADM cassettes is in progress and will allow for virtually any gene to be studied in the future, providing an unparalleled platform for the study of developmental lineage relationships in combination with functional genetic analysis.
Protocol
Mouse protocols were reviewed by institutional preclinical core facility (PCF) and internal ethical committee at IST Austria. All breeding and experimentation were performed under a license approved by the Austrian Federal Ministry of Science and Research in accordance with the Austrian and EU animal laws.
1. Breeding of experimental mice for MADM clonal analysis
- Set up timed experimental MADM matings (>P56; CD-1) in the late afternoon (5:00 PM) and check for vaginal plugs the proceeding morning (8:00 AM). The morning the plug is present counts as day 0.5. See Figure 2 for an overview of the experimental mouse mating setup. Ensure that timepoints for TM induction of CreERT2 activity and analysis are appropriate to address experimental questions.
NOTE: For more information refer to Figure 3 and representative results below. - For postnatal sampling, set up breedings to generate foster mothers in parallel.
NOTE: These should be started up to 1−2 days prior to setting up experimental breedings.
2. TM induction in MADM mice
- Prepare a 20 mg/mL TM working solution by dissolving it in corn oil in a 15 mL or 50 mL conical centrifuge tube and placing it on a rocking platform for ~4 h at room temperature (RT), ensuring TM is completely dissolved. Store working solution at 4 °C covered with aluminum foil and use within 2 weeks.
- To induce MADM recombination events, deliver a single injection of TM intraperitoneally (IP) using a 1 mL tuberculin syringe and a 25 G needle into a timed pregnant dam. Depending on the stage of cortical neurogenesis, inject TM between E10−E15 at a dose of 1−2 mg/pregnant dam. For early timepoints (i.e., E10) use a maximum of 1 mg/pregnant dam (25 mg/kg) to prevent complications during pregnancy11. For timepoints between E11-E15 use 2 mg/pregnant dam (50 mg/kg)7.
NOTE: Alternatively, TM can be administered with an oral gavage for late pregnancies. - For MADM clonal analysis to postnatal timepoints, recover live embryos at E18−E19 through Caesarean section, and then raise pups with a foster mother.
NOTE: Depending on the health status of the pregnant female, it may not be necessary to perform a Caesarean section but raising pups with a foster mother is still required because the original TM-treated mother may have trouble lactating. - To recover live embryos by Caesarean section or to retrieve embryonic timepoints for analysis, sacrifice the pregnant dam by cervical dislocation.
- Place the animal in a supine position and disinfect fur with 70% ethanol. Make a small incision in the skin in the lower abdomen above the uterus using surgical forceps and scissors. Make a second incision through the muscles and the abdominal muscular wall to reveal the peritoneum.
- Remove the uterus by separating from the surrounding tissues with scissors. Transfer intact uterus onto a glove filled with warm water to increase the embryo survival rate until each is removed from the amnion individually.
- Use fine tipped scissors and fingers to carefully open the uterine walls to release embryos. Do not cut the umbilical cords too close to the body to prevent extensive blood loss. If embryos are to be used for analysis, proceed to step 3.9. If pups are to be fostered, proceed to step 2.8.
- If fostering is required, clean the pups before transferring them to the foster mother. While cleaning the pups, gently press the chest from time to time to initiate breathing. Place back on a second glove filled with warm water to improve survival rate.
NOTE: It is important to gently remove any remaining amnion and/or placenta with a paper towel. - Before transferring pups to the foster mother, remove the foster mother from her cage, remove the original pups, and replace with the experimental pups. Return the foster mother to her cage.
NOTE: See discussion for additional suggestions to improve fostering acceptance rates. - If genotyping is required, collect toe or tail biopsies between P6−P8.
NOTE: Perform this step only if animal experimental licenses approve this practice.
3. Tissue preparation for MADM clones in the brain
NOTE: For experiments that include postnatal tissue (≥P4), proceed to step 3.1. For embryonic timepoints and early postnatal (P0−P3), continue to step 3.9.
- Anesthetize the experimental MADM animal with an IP injection of a ketamine/xylazine/acepromazine solution (65 mg, 13 mg, and 2 mg/kg body weight, respectively) and confirm that the mouse is unresponsive by pinching the hind paw.
NOTE: Both male and female MADM mice (CD-1 background) are used for analysis. If genotyping is required, collect ear biopsies at this point. - Place the anesthetized animal in the supine position on the perfusion surgery tray and disinfect fur with 70% ethanol. To begin surgery, carefully make an incision with scissors and surgical forceps through the outer layer of the skin and then a second incision through the muscle layer. Lift the tip of the sternum and cut connective tissue on the sides, taking extra caution to avoid cutting the liver. The thoracic cavity will be visible.
- Snip the diaphragm and lift to reveal the heart. Carefully trim the rib cage and pin to the surgical tray to expose the heart. For pups, remove the rib cage completely.
- Insert a needle with phosphate buffered saline (PBS) into the lower left ventricle (paler tissue). Using small iris scissors make an incision to the posterior end of the right atrium (dark red tissue) for the blood to drain.
- Perform perfusion with PBS followed immediately by freshly made, ice-cold 4% paraformaldehyde (PFA) prepared in PBS. For pups (P4−P10) use syringes to perform perfusion. Fill one syringe with 10 mL of PBS and another with 10 mL of 4% PFA. Ensure all air bubbles in the syringes have been removed. For older animals, use a peristaltic pump.
- Begin perfusing with PBS (10 mL at 2−4 mL/min in pups; 20 mL at 4−6 mL/min for adults using a peristaltic pump). The liver will become clear and pale yellow if the needle is positioned correctly.
- Once complete, remove the needle from pups and insert the needle containing PFA into the same hole. For adults, stop the peristaltic pump before exchanging the PBS solution with ice-cold PFA, making sure to avoid bubbles in the uptake tubing. Resume perfusing with PFA (10 mL at 2−4 mL/min in pups; 30 mL at 4−6 mL/min for adults using a peristaltic pump).
- When perfusion is complete, decapitate the mouse and remove the brain through careful dissection. Transfer brain to 4% PFA. Use at least 5x the brain volumes (i.e., ~5−10 mL of PFA in a 15 mL conical centrifuge tube) and incubate overnight at 4 °C for postperfusion fixation to ensure complete fixation of tissue. Continue to step 3.10.
- For embryonic tissue and early postnatal tissue (i.e., P0−P3), after performing a Caesarian section, decapitate the embryos with scissors. If genotyping is required, collect the tail of the embryo at this point. Immediately dissect out the brain and transfer to a 12 well plate containing 2−3 mL of 4% PFA/well. Incubate overnight at 4 °C for postfixation.
- Next morning exchange PFA with 10 mL (adult) or 2−3 mL (embryo) of PBS and repeat wash 3x for 15 min at RT. Transfer tissue to 30% sucrose solution in phosphate buffer (PB) and store at 4 °C on a rocking platform until the tissue sinks in the solution.
- Embed the brain in optimal cutting temperature (OCT) compound in an embedding mold, taking care to orient the brain for either coronal or sagittal sectioning. Freeze by placing the embedding mold on dry ice until OCT becomes completely opaque (~10−15 min). Store tissue at -80 °C until further use.
4. Preparation of MADM tissue for immunohistochemistry
- Attach the tissue block to the specimen disk in the cryostat by applying a ring of OCT to the disk and placing the block directly into the OCT when it begins to freeze. Ensure the block is correctly oriented for the desired cutting plane.
NOTE: Here, coronal sectioning to investigate cortical MADM clones is described in detail. - Set the block temperature in the cryostat to -20 °C and the blade temperature to -21 °C.
- Allow the tissue block to adjust to the chamber temperature by mounting the specimen disk to the specimen holder and leave in cryostat for ~5 min before beginning sectioning.
- Trim block in thick sections (45−60 µm) until the tissue region of interest is reached.
- Once the edge of the cortex is clearly visible, stop sectioning and lock the blade. Ensure the blade is shielded before trimming the block.
- Trim excess OCT surrounding the tissue with a blade, leaving ~1−2 mm of OCT on all sides of the brain.
- Next orient the block so that one of the lateral edges of the cortex is oriented downward and the other upwards (i.e., the most rostral edge of the cortex is pointed right).
- Begin sectioning with a thickness of 45 µm for adult clones and 30 µm for embryonic clones. Perform each section individually and use a small brush to keep the area below the knife clean of any debris left while trimming the block.
NOTE: If this is not done and a section falls, it may be difficult to determine the correct order of the slices. - If sections begin to curl, trim the edges of the block and/or carefully adjust the glass antiroll plate.
- For embryonic clone analysis, mount sections directly to a frosted slide. Dry on a heating plate at 37 °C before proceeding directly to step 5.6.
NOTE: Several sections can be added to one slide, but ensure the sequential order is maintained. - To collect adult clones, prepare 24 well plates containing 1 mL of PBS/well (typically, 5−6 plates per brain). Starting from the first well, with cold forceps collect individual serial sections in PBS in the order of sectioning.
NOTE: The floating section method is adopted for adult tissue to ensure that no sections are missed and that mounted sections contain no wrinkles. - Stop sectioning once the end of the neocortex is reached.
- For adult clones, proceed to mounting floating sections.
NOTE: Sections can be kept in PBS at 4 °C for up to 24 h.
5. Mounting adult tissue for imaging
NOTE: The following tools are required: small paint brush, Petri dish, PBS with 0.5% Tween (PBS-T), adhesion slides (Table of Materials), mounting medium (Table of Materials), coverslips, and forceps.
- Fill a Petri dish with PBS-T.
NOTE: Detergent is used to aid in the mounting process. If staining for additional antigens that are sensitive to detergents (i.e., glycoproteins) is necessary, it is best to skip the addition of Tween. - Place an adhesion slide into the PBS-T so that it is nearly covered up to the label.
- Transfer the first section into the PBS-T.
- Using a small paint brush, maneuver the section onto the slide and arrange it to preserve the order of cutting. Proceed in the same way with all further sections.
- Once all sections are in position, place the slide (~12−16 sections/slide) in a dark slide chamber. Slightly lift the lid to allow the sections to dry completely (~10−20 min), ensuring that they remain adhered in subsequent steps.
- If performing immunohistochemistry for additional antigens, proceed directly to section 6 or 7.
NOTE: For embryonic timepoints, it is necessary to perform immunostaining steps for at least GFP and tdT (section 6). For adult clones, this is only required if staining for additional antigens in parallel (sections 6 and 7). - Rehydrate and wash sections 1x with 1x PBS for 5 min to remove residual PBS-T. Apply ~1 mL of 4′,6-diamidino-2-phenylindole (DAPI) diluted in 1x PBS (1 μg/mL) to the slide, ensuring all sections are covered and incubate for 15 min.
- Carefully remove the DAPI and wash 1x with 1x PBS for 5 min. Remove excess PBS and dry for ~1−2 min before embedding in 110 µL of mounting medium. Seal with a 24 x 60 mm coverslip and let dry for at least 3 h before imaging.
6. Immunostaining for GFP and tdT only
NOTE: This section is necessary for embryonic clones.
- Place slides horizontally in a humidified slide incubation chamber. Mark slide boundaries with a wax marker to minimize the amount of buffer required.
- Rehydrate sections with 1x PBS. To improve staining quality, work with freshly sectioned tissue.
- Add 250−400 µL of blocking buffer (0.5% Triton X-100, 2−3% normal donkey serum in 1x PBS) per slide, ensuring all sections are covered. Incubate for 1 h.
NOTE: The concentration of detergent (Triton X-100 or Tween-20) will vary depending on the additional primary antibodies used because some antigens are more sensitive to detergents than others. - Remove blocking buffer and add primary antibodies in blocking buffer to the slide (300−400 µL/slide).
NOTE: An example of a standard primary antibody reaction for anti-GFP/anti-tdT (MADM) could use chicken anti-GFP (1:500) and rabbit anti-RFP (1:500). - Incubate with primary antibodies overnight at 4 °C.
NOTE: The slides must be incubated perfectly horizontally with buffer covering all sections. Otherwise, uneven or poor staining may result. - Confirm the next morning that the blocking buffer with primary antibodies still covers all sections on the slide. If not, repeat the incubation step for 3−4 h at RT.
- Remove primary antibodies and wash 4x with 1x PBS for 10 min at RT.
- Add secondary antibodies diluted in blocking buffer to slide (300−400 µL/slide): Alexa Fluor 488 anti-chicken IgG (1:500) and Cy3 anti-rabbit IgG (1:500).
- Incubate at RT for 2 h. Keep slides covered from light to prevent bleaching of fluorophores.
- Remove secondary antibodies and wash 2x with 1x PBS for 10 min.
- Incubate with DAPI diluted in PBS (1:5,000) for 15 min.
- Wash sections 1x with 1x PBS for 10 min.
- Remove excess PBS and dry for ~1−2 min before embedding in 110 µL of mounting medium.
- Seal with 24 x 60 mm coverslip and let dry for at least 3 h before imaging. Image slides within 1−2 weeks after performing immunohistochemistry to ensure optimal signal.
7. Immunostaining for GFP, tdT, and additional antigens
- Perform steps 6.1−6.3.
- Remove blocking buffer and add primary antibodies in blocking buffer to the slide (300−400 µL/slide).
NOTE: When staining for three or more antigens (i.e., GFP, tdT, and a protein of interest) and the antibody for the protein of interest was raised in rabbit, it is recommended to use the anti-tdT (goat) primary antibody at a dilution of 1:500. An example of a primary antibody reaction for three antigens with alternative tdT staining could use chicken anti-GFP (1:500), goat anti-tdT (1:500), and antibody against the protein of interest (i.e., rabbit). - Perform steps 6.5−6.7.
- Add a secondary antibody mix diluted in blocking buffer to slide (300−400 µL/slide): Alexa Fluor 488 anti-chicken IgG (1:500), Cy3 anti-goat IgG (1:500), and Alexa Fluor 647 anti-rabbit IgG (1:500).
- Perform steps 6.9−6.14.
8. Confocal image acquisition and quantification of MADM clones
- Identify and document brain sections containing clones and their locations in the cortex.
NOTE: The number of sections a clone spans will vary depending on when the clone was induced, the CreERT2 driver, and the time of analysis. This step can be performed on either a confocal microscope or an epifluorescence microscope. - Using an inverted confocal microscope, begin by selecting and configuring the correct laser lines and filters. For MADM brains, select DAPI, GFP, and tdT (excitation: 358 nm, 488 nm, and 554 nm, respectively; peak emission: 461 nm, 507 nm, and 581 nm, respectively). Ensure the pinhole is set to 1 airy unit for optimal imaging quality.
- For confocal specific settings, image clones with a 20x objective and 1x zoom. For images that will be used in quantifications, use a scanning speed pixel dwell value of 1.52−2.06 µs (values 7−8 in the image acquisition software) with no averaging. Adjust laser intensity and gain settings for each channel as appropriate.
NOTE: Depending on the required image quality, settings for scanning speed and averaging may vary. - Once the clone is clearly identified, arrange imaging tiles to cover all relevant sections in the clone. Adjust the z-stack so that all MADM labeled cells in the clone are captured with an interval of 1.5 µm/z-stack slice. Adjust the tiled region so that the entire width of the cortex is captured when imaging the clone (i.e., from pial surface to the corpus callosum).
- Image individual clones spanning multiple sections consecutively, ensuring that any sections without cells within a clone are still imaged for the purpose of 3D reconstruction and correct interpretation of cell spatial information.
- Analyze each section containing cells of a MADM clone sequentially from the rostral to the caudal end of the cortex. Distinguish individual neurons and glia based on their morphology and/or marker staining. Record positional information in parallel based on respective layer boundaries defined by nuclear staining (DAPI).
NOTE: See Figure 4 for representative results for embryonic analysis and Figure 5 for representative results for adult analysis.
9. Serial 3D reconstruction of clones
NOTE: The 3D reconstruction of individual clones imaged over serial brain sections is useful for visual display as well as the analysis of 3D clonal architectures and can be performed according to the following steps.
- Stitch and fuse confocal tiled images based on acquisition parameters using image acquisition software. Open .czi file and then run the Stitching method under the Processing tab in the ZEN software (Zeiss).
- Export stitched image stacks as individual z-planes in TIFF format. Open stitched .czi file and then run the Image Export method under the Processing tab. For multichannel images, export as Red/Green/Blue images for subsequent image processing.
- Repeat steps 9.1 and 9.2 for each serial brain section of a clone.
NOTE: For accurate 3D reconstruction, all brain sections within a clone, including the ones without labeled cells, must also be processed. - Concatenate individual images into a single stack in order beginning from the most rostral to the most caudal z-plane using open source image processing software such as ImageJ/Fiji67,68.
NOTE: Any blank images at the edges of each brain section should be removed at this point. - If required, correct the image stack obtained from step 9.4 for misalignment using an ImageJ plugin called "MultiStackReg" by following steps 9.5.1−9.5.5. If image alignment is not required, proceed to step 9.6.
NOTE: This plugin performs image alignment of the channel with highest contrast (usually DAPI) and then applies the recorded transformation to the other channels, thus allowing reliable image alignment of multichannel stacks. An auxiliary plugin called "TurboReg" must be preinstalled.- In ImageJ, install the "MultiStackReg" and "TurboReg" plugins.
- Open the image stack of clone images obtained from step 9.4 to be aligned. Split channels into DAPI (blue), GFP (green), and tdT (red) within Color option under Image tab.
- Run "MultiStackReg" plugin to align DAPI channel by "Rigid Body" transformation and save the transformation file.
- Apply the saved transformation file to the other two channels using "MultiStackReg".
- Merge all three aligned channels and save aligned stack.
- To orient the clone in ImageJ rotate stack of clone images obtained from step 9.4 (or step 9.5.5 after alignment) in a vertical orientation with the pial surface on top and the corpus callosum at the bottom. Crop in the xy plane if necessary.
- For both qualitative presentation and quantitative analysis, generate a maximum z-projection image (step 9.8) or perform 3D rendering (step 9.9) of the clone.
- In ImageJ, open the image stack from step 9.6 and select Z-projection option with projection type Max intensity. This will generate an image of the entire clone projected on the same plane.
- In ImageJ, open the image stack from step 9.6 and select 3D Project z-function to generate a 3D visualization of the clone that can be rotated.
NOTE: It is important in this step to input the correct slice interval equivalent to the thickness of individual z-stacks during image acquisition. The interpolation tool should be used to remove gaps between slices.
Results
MADM results in the reconstitution of functional green and red fluorescent proteins with two daughter cells each expressing one of the two fluorescent proteins upon G2-X chromosome segregation events (Figure 1A). Because MADM events result in permanent and distinct labeling of the two descendent lineages, quantifiable assessment of green and red daughter cell lineages (subclones) can be performed. Variables including division pattern (e.g., symmetric versus asymmetric) and potential (e.g., the number of progeny) of the original progenitor can be determined. Quantifying each fluorescently labeled subclone is informative when retroactively determining if the original progenitor cell is undergoing symmetric proliferative divisions, or asymmetric, neurogenic divisions at the time of TM induction. Previous studies grouped Emx1-CreERT2 or Nestin-CreERT2 derived excitatory projection clones in the cortex into two broad classes7,11,46. The first, termed "symmetric proliferative clones", are composed on average of a considerable number of neurons, with both green and red subclones containing four or more neurons each. The second group, "asymmetric clones" defines a class of clones where the "minority" subclone contains fewer than three neurons and the "majority" subclone, four or more11. These definitions are specific to cortical RGPs and may need to be revisited for other brain regions and tissues. For both classes of cortical clones, progeny will be distributed throughout the superficial and deep layers.
When designing MADM clonal studies there are a number of aspects that must be taken into consideration. The time when MADM events are induced by administration of TM is a key consideration (Figure 3). For cortical excitatory projection neuron MADM clones (i.e., using Emx1-CreERT2 or Nestin-CreERT2) at E10, nearly all RGPs were still undergoing symmetric divisions11. Therefore, induction at E10 with TM captured multiple rounds of proliferative RGP amplification and resulted in clones with high neuron numbers. However, the number of RGPs at E10 was generally small and thus TM administration generated very few MADM events (sometimes less than one per brain). The majority of RGPs switched from symmetric to asymmetric neurogenic divisions at around E12. To target strictly asymmetric neurogenic clones, it was best to induce at E12 or later (Figure 3). The time between TM induction and observing MADM recombination events in the cortex tended to be less than 24 h. IP injections were the preferred method for administering TM at embryonic stages for this method because it led to greater reproducibility in clonal induction. It is also important to keep the TM dose to a minimum for two reasons. First, if the MADM recombination rate increases, the probability of inducing multiple, perhaps overlapping, clones is higher. Second, if too much TM is delivered, an increased rate of abortion, embryo reabsorption, and smaller litter sizes may be observed. Abortions in approximately half of all pregnant dams was observed when TM injections were delivered at E10. This frequency decreased from E11 onwards and diminished to approximately 1/3 of pregnant dams aborting. For a summary of TM doses, induction times, and CreERT2 drivers used in previous MADM studies, refer to Table 1. Reporter activity in the absence of TM was observed with some TM-inducible CreERT2 drivers69. Ectopic expression or MADM recombination events in the absence of TM was not observed with the Emx1-CreERT2 of Nestin-CreERT2 drivers. This may be partially due to the fact that TM-mediated chromosomal trans-recombinations occur at approximately 1:1,000 to 1:10,000 lower frequency than cis-recombinations, reducing the probability of ectopic MADM labeling.
Another factor to consider when planning a MADM clonal analysis experiment is the duration of the study. Varying the length of the time between TM induction and when the experiment was analyzed (A) (time window) displays stem cell dynamics over time64. Short embryonic time windows (i.e., TM/E11−A/E13; TM/E11−A/E16) captured the dynamics of embryonic neurogenesis (Figure 4). Comparing clones from two or more time windows provides quantitative insight into the number of cells produced and how neuron distribution varies at different stages of lineage progression64. To capture the entire potential of individual clones, it is necessary to extend the time window analyzed into postnatal or adult timepoints7,11,12. Examples of neocortical clones induced in the embryo and analyzed in the adult are shown in Figure 5. Of note, cortical neurogenesis is mostly completed and gliogenesis increases by E17. Approximately 1/6 neurogenic RGP also proceed to generate astrocytes and/or oligodendrocytes11.
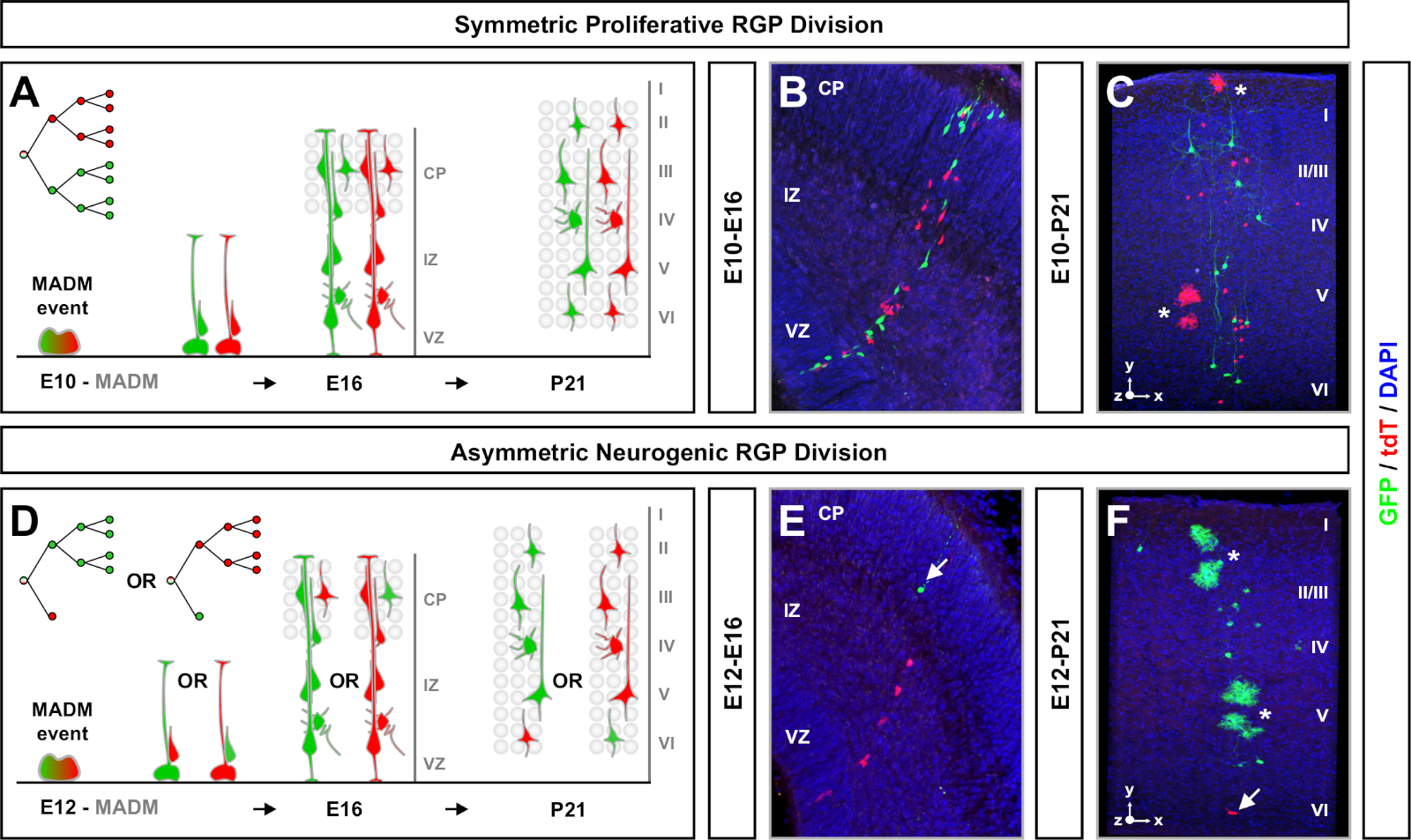
Symmetric clones occur when RGPs undergo one or more rounds of proliferative division11. RGP clones induced between E10−E12 were on average larger in size and provided more spatial features of the final neuron distribution (Figure 4A-C). Clones with neurons relatively equally distributed throughout deep and superficial layers took on a "cylinder" shape while clones with neurons more dispersed in superficial layers than deeper layers developed a "cone" shape11. To fully capture the spatial and morphological information of a clone, it was necessary to computationally reconstruct each clone using sequential images. To measure clonal dispersion, the maximal lateral dispersion (measured in all dimensions) in superficial layers (LII−VI) of a clone was compared to neuron dispersion in deep layers (LV/LIV). This ratio (distribution upper:distribution lower) provided a quantifiable readout of the overall clone shape.
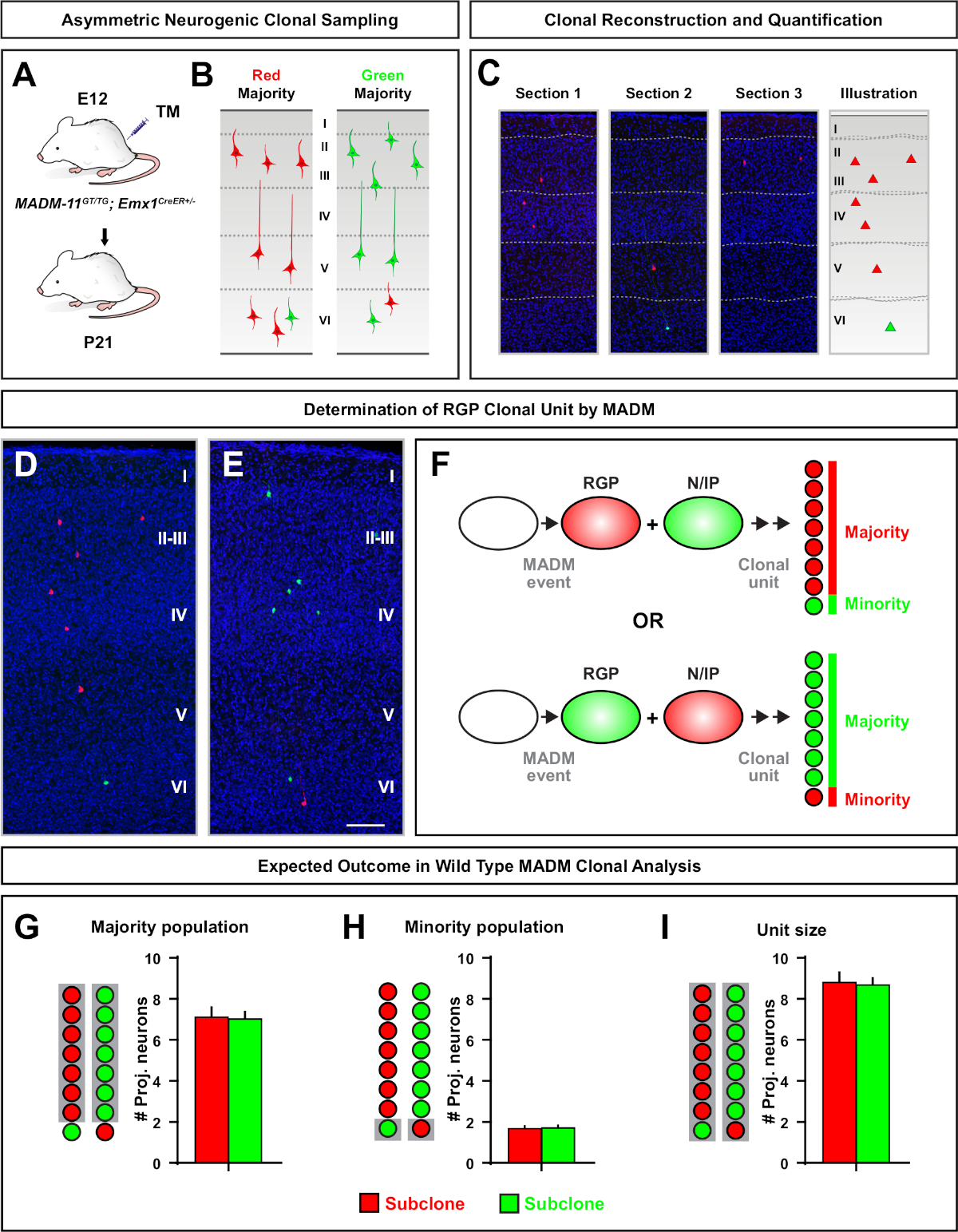
Asymmetric clones, where the minority subclone was three or less, provided insight into the neuronal output of a single RGP (Figure 4D-F and Figure 5A-F)7,11,12. The majority population (large subclone) could be labeled either in red or green, with an average of approximately seven excitatory projection neurons per clone when induced using an Emx1-CreERT2 or Nestin-CreERT2(Figure 5G)7,11,12. The total number of cells in a MADM clone could be further dissected by analyzing the distribution of neurons in the large subclone across superficial and deep layers. The minority population (small subclone) was labeled by the reciprocal color and was on average 1−2 cells per clone (Figure 5H). The total "unit size", which was on average 8−9 neurons, could be calculated by adding the small and large subclones together (Figure 5I)7,11,12. It is important to note that while the neuronal output of RGPs was highly predictable, there was a degree of clonal heterogeneity12,70.
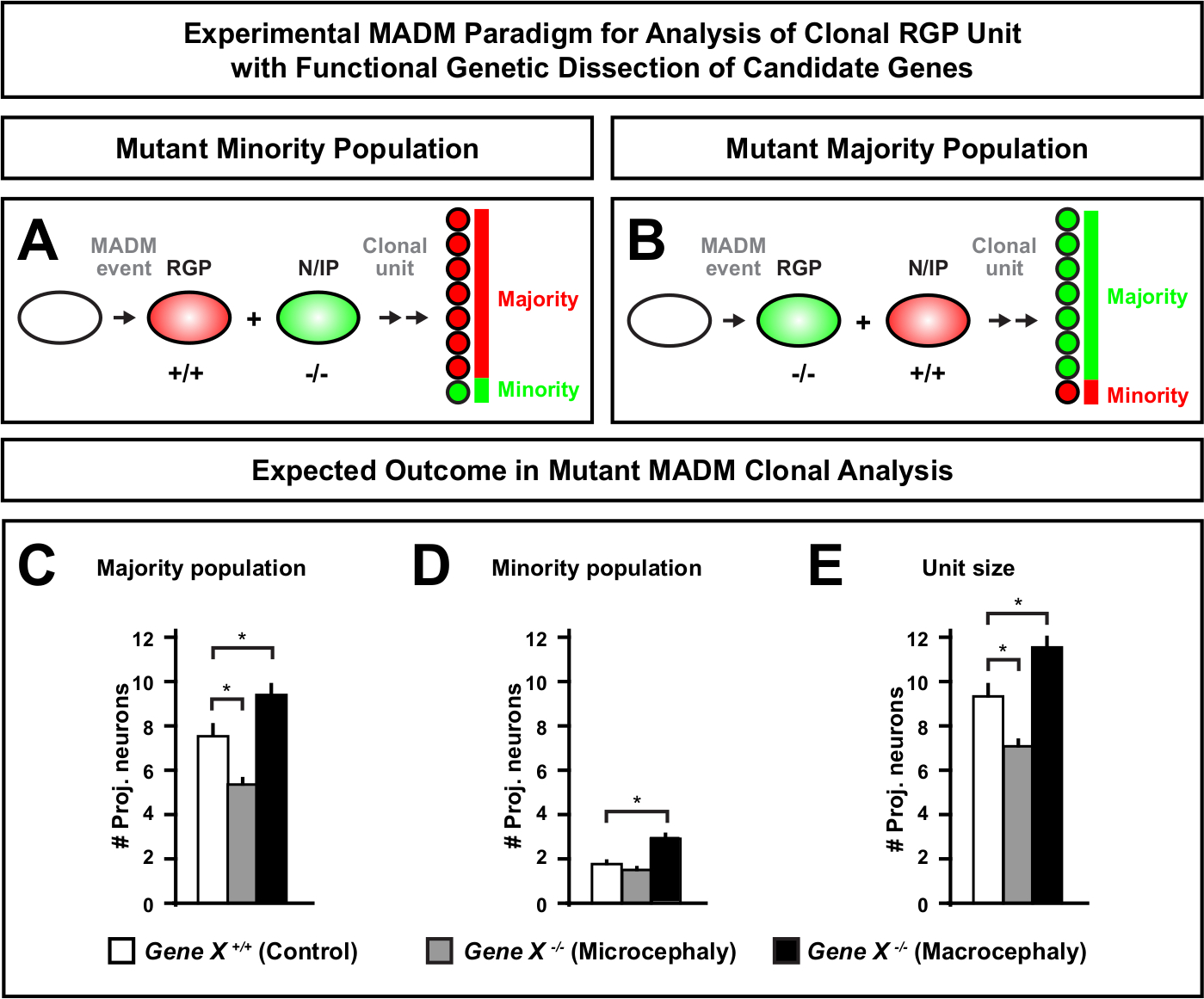
Introduction of a mutation distal to the MADM cassette enables the generation of genetic mosaics, providing a unique method to dissect the molecular regulators of stem cell lineage progression. As such, MADM provides an unparalleled experimental platform to study the cell-autonomous function of a gene (e.g., its association to microcephaly or macrocephaly). By comparing clones induced in a MADM genetic mosaic to clones induced in a control MADM, a highly quantitative readout of changes in neuron numbers and distribution can be generated. Previous MADM-based studies quantified the cell-autonomous function of Otx1 in microcephaly formation at the clonal level (see Figure 6A-E for a representative example)11. In another study, MADM clonal analysis demonstrated that Ndel1 does not cell-autonomously regulate projection neuron numbers, but instead the ability of newborn neurons to enter or migrate within the cortical plate, which later forms the adult cortex46. These studies demonstrated the highly quantitative nature of MADM clonal analysis in studying the cell-autonomous functions of genes regulating cortical development. There are currently no examples in the literature using MADM to study genes implicated in macrocephaly at the clonal level. However, in future studies analysis of genes relevant to the control of cortical size in general can provide highly desirable insights at the molecular and cellular level.

Figure 1: The MADM principle for lineage tracing and clonal analysis at single stem cell level. (A) To perform lineage tracing and clonal analysis with MADM, two components must be present. First, MADM cassettes must be targeted to identical loci on homologous chromosomes. Cassettes consist of two chimeric fluorescent reporter genes, eGFP (green, [G]) and tandem dimer Tomato (red, tdT[T]). The GT cassette contains the N-terminus of eGFP and the C-terminus of tdT, separated by an intron containing a loxP site. The TG cassette is constructed inversely, with the N-terminus of tdT and the C-terminus of eGFP. Second, the expression of Cre recombinase must occur in the cell containing the targeted MADM cassettes. The loxP sites serve as a target for Cre-mediated interchromosomal recombination, resulting in the reconstitution of both expression cassettes simultaneously. If recombination occurs during the G2 phase of the cell cycle followed by X segregation (G2-X), the two daughter cells will express one of the two fluorescent proteins. (B) MADM principle for genetic mosaic analysis at a single clone level. Mutant alleles (point mutations, deletions, insertions, loxP-flanked conditional alleles as depicted in Figure 1B, etc.) can be introduced distal to the TG-MADM cassette via meiotic recombination (see Figure 2 and Hippenmeyer et al.46 for details on how to introduce mutant alleles into the MADM system). If a G2-X Cre recombinase-mediated interchromosomal trans-recombination occurs between MADM cassettes it results in one GFP+ homozygous mutant cell (GeneX-/-) for the gene of interest and one tdT+ homozygous wild type cell (GeneX+/+) in an unlabeled heterozygous environment46,47,71. Alternate labeling outcomes not used in the clonal analysis (i.e., yellow cells) have been previously described in detail11,46,47. Please click here to view a larger version of this figure.
{kind=link}

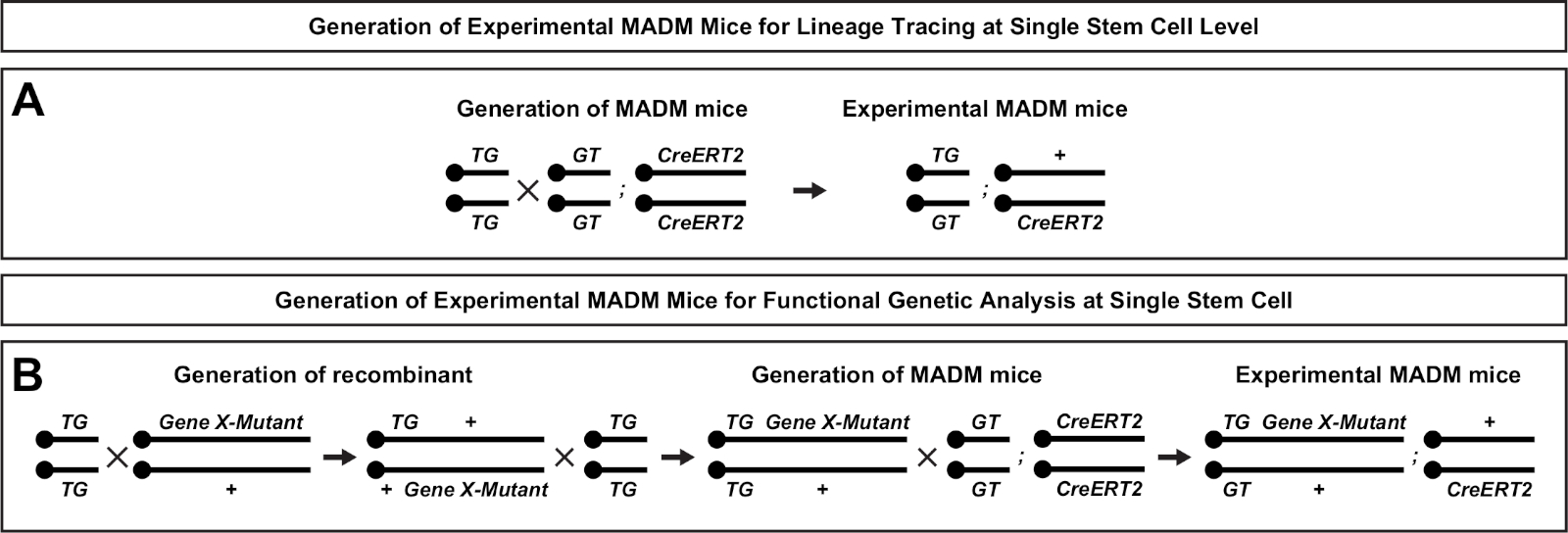
Figure 2: Breeding schemes for generation of experimental MADM mice for lineage tracing. Breeding scheme for the generation of control MADM (A) and Gene X MADM (B) experimental MADM mice for clonal analysis. For more information regarding MADM breeding paradigms see Beattie et al.7 and Hippenmeyer et al.7,46. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Time course paradigms for MADM-based clonal lineage analysis. Schematic of the experimental design time windows. For longitudinal sampling paradigms, the timepoint of clone induction remained constant and the length of time before analysis varied. In progressive interval sampling, the timepoint of analysis remained constant, but the time of induction varied. A combination of one or both approaches can be used depending on the questions addressed. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: MADM clonal analysis in the developing and adult neocortex. TM-mediated MADM clone induction in symmetrically proliferative (TM at E10) (A-C) and asymmetrically neurogenic (TM at E12) (D-F) dividing RGPs. Depicted are individual MADM clones in vivo in the developing (TM/E10−A/E16 and TM/E12−A/E16) (B,E) and adult (TM/E10−A/P21 and TM/E12−A/P21) (C,F) in MADM-11GT/TG; Nestin-CreERT2+/- (B,E) and MADM-11GT/TG; Emx1-CreERT2+/- (C,F). Neuron output was independent of subclone color and green majority/minority subclones could be compared to red majority/minority subclones under control conditions7,11. Approximately 1/6 of adult clones also contained astrocytes and/or oligodendrocytes, indicated by white asterisks. Panels B and F are reproduced with permission from Hippenmeyer et al.46 and Rulands and Simons72, respectively. CP = Cortical plate. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: MADM clonal analysis to quantify RGP-mediated neuron output. Analysis of excitatory neuron (unit) production by individual neurogenic RGPs at the clonal level using MADM7,11. (A) Experimental paradigm for inducing mostly asymmetric MADM clones in the developing cortex. (B) Possible asymmetric clone outcomes with the majority subclone labeled in either green or red (C) Representative consecutive sections spanning a single neurogenic asymmetric clone (D,E) 3D reconstruction images of representative asymmetric G2-X MADM clones with majority population in red (D) or green (E) in MADM-11GT/TG; Emx1-CreERT2+/- with TM induction at E12 and analysis at P21. Note both green and red labeled cells are wild type. (F) Schematic indicating the two possible experimental MADM clone outcomes. (G) Quantification of the size of the majority population arising from renewing RGPs in MADM-11 clones. (H) Quantification of the size of the minority population arising from renewing RGPs in MADM-11 clones. (I) Quantification of the unitary size of asymmetric neurogenic MADM-11 clones. Hypothetical values could represent mean ± SEM. Scale bar = 100 µm (D and E). TM = Tamoxifen. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: MADM clonal analysis to study genes that lead to microcephaly and macrocephaly. Hypothetical MADM clonal analysis results when performing functional genetic dissection of candidate genes that lead to microcephaly or macrocephaly. To dissect the cell-autonomous functions of a gene of interest (Gene X) on neuron output, MADM requires mutant alleles to be introduced distal to the MADM cassettes via meiotic recombination (for details how to introduce mutant alleles into the MADM system see also Figure 2, Hippenmeyer et al.46, and Laukoter et al.46,73). (A,B) Schematic indicating experimental MADM paradigm for functional analysis of clonal RGP units. The mutant subclone can either form the minority (A) or majority (B) population. (C-E) Hypothetical MADM clonal analysis results when quantifying control MADM (white bars), Gene-X MADM microcephaly (gray bars) and Gene-X MADM macrocephaly black bars) asymmetric clones. (C) Quantification of the size of the majority population. (D) Quantification of the size of the minority population. (E) Quantification of the unitary size of asymmetric neurogenic clones. Hypothetical values could represent mean ± SEM. S = Hypothetical scenario where difference in subclone cell number could reach significance, relative to control. Please click here to view a larger version of this figure.
{kind=link}
Table 1: MADM clonal studies in the literature. Summary of studies in the literature containing MADM clonal lineage experiments, including CreERT2 driver used, TM dose, and time of injection. Please click here to view this table (Right click to download).
Discussion
A method to use MADM to track cell lineage of individual RGPs in vivo in the developing neocortex is described. When combined with TM-inducible CreERT2, MADM events can be precisely timed, providing a highly qualitative and quantitative visual readout of stem cell division patterns at the single cell level. By titrating the dose of TM delivered, in an ideal situation an average of less than one clone per cortical hemisphere can be obtained, providing adequate spatial separation to unambiguously distinguish individual clones. By maintaining tissue integrity, this method also captures essential information regarding position, morphology, and absolute cell numbers. MADM cassettes on Chr. 117,11,12,46,56,57, on Chr. 751, and the original MADM at Rosa2647,53,59 have been used in MADM clonal analysis studies. The high-resolution of individual cells provides unprecedented insight into both morphology and the clonal relationship of daughter cells and permits the live imaging of proliferating stem cells and emerging clones46,52.
Caesarean section and fostering of pups for analysis of clones at postnatal timepoints is a necessary and critical step in the protocol. Depending on the health status of the TM-treated pregnant dam, it may not be necessary to perform a Caesarean section. However, raising the pups with a foster mother is still required, because the TM-treated mother may have trouble lactating. No differences have been observed in the need for fostering with different CreERT2 drivers. Both MADM lines and foster mothers are maintained on an outbred CD-1 background. If Caesarean section is not necessary, the TM-treated pregnant dam used to generate experimental pups may be reused for additional experimental breedings in accordance to principles of 3R (note that this can only be done if animal experimental licenses approve of this practice). Foster mothers can be used for fostering pups within 2 days after they give birth, but higher success rates have been observed when foster mothers give birth on the same day as the experimental mice that must be fostered. Therefore, it is important to set up timed matings for foster mothers in parallel to experimental matings in step 1.1. Maintaining a similar litter number as the original foster mother's litter can improve the survival rate of fostered pups, and therefore removal of some to all of the original litter may be necessary. Additional steps that may improve fostering includes rubbing the experimenter's gloves with litter and food (to remove the scent of the gloves); rubbing the pups gently after the Caesarean section with fragments of the foster mother's soiled litter and nest; and placement of the pups in close contact with the foster mother's pups prior to their placement in the foster mouse cage.
As in other reporter-based lineage tracing methods, careful consideration must be taken when choosing the optimal CreERT2 driver for MADM clonal experiments. First, the promoter used must express the recombinase both temporally and spatially in the progenitor population of interest. Finding this promoter can be challenging, because some promoters may change expression patterns or become silenced at different stages of development. To improve cell type specificity multiple site-specific recombinases, each driven by separate promoters, have been used. When one or both recombinases are expressed in the same cell, this labels the cell and its progeny with a fluorescent reporter74,75,76,77. In summary, it is important to choose a CreERT2 driver that is specific to the population of progenitors being analyzed.
The most critical step in this method is the identification of a clone, because all cells must be derived unambiguously from a single recombination event (step 8.1). Titration of TM concentration ensures less than one cluster of red/green cells per brain hemisphere and maximizes the probability of analyzing a single clone (step 2.2)7,11. Clones should be discarded if neighboring clusters of cells occur within 500 µm of the clone of interest. Therefore, it is important to examine several sections before and after the appearance of a clone to ensure that there are no additional recombination events nearby. Due to weaker signal of the fluorophores, it is necessary to perform immunohistochemistry for eGFP and tdT in embryonic clones (see section 6). This is only recommended in adult clones if additional antigens will be colabeled. When imaging clones, it is important to capture the entire width of the cortex where the clone is located (i.e., from pial surface to the corpus callosum; see step 8.4) to not miss any cells. This also facilitates image alignment during image processing (section 9). Section 8 of the protocol requires an inverted confocal microscope but can be adapted depending on the microscope setup available. Epifluorescence microscopy can be used, but confocal microscopy is recommended because this leads to a decrease in light contamination from outside the focus plane. It is also important that the laser intensity and gain is adjusted so that green, red, and yellow cells can be unambiguously identified. Regardless of the setup, it is recommended to use an objective of at least 20x to ensure full spatial separation of closely positioned cells. In addition to recording the cortical depth of all cells (step 8.6), cortical regions where the clones are located must be identified using a brain atlas such as the Allen Brain atlas or other stereotaxic coordinate maps. A file naming paradigm should also be adopted to make sure clone images are easily identifiable. The following information could be included in the file naming: unique image ID, date image was taken, genotype of animal, age of induction, age of analysis, image number in relation to the rest of the images from the same clone.
Introduction of a mutation distal to one MADM cassette distinctively allows the generation of genetic mosaics71 and permits the dissection of molecular regulators of lineage and cell type diversity at the clonal level7,11,46,62. To generate a genetic mosaic with MADM, the MADM cassettes must be meiotically linked to the same chromosome as the gene of interest (see Figure 2 for breeding scheme). This limits current clonal analysis with MADM to genes located on Chr. 751, Chr. 1146, Chr. 1251, and Chr. 6 distal to the Rosa26 locus47. Future studies will use MADM cassettes targeted to any chromosome, permitting the mosaic analysis of virtually all genes of the mouse genome at the clonal level.
Finally, MADM is not limited to the analysis of progenitor cells in the developing neocortex. The study of many stem cell niches could benefit from the ability to resolve spatiotemporal arrangements of clonally related cells. By applying MADM to other regions of the brain, disease conditions (e.g., cancer), or in other tissues47,50,51,52,53,54,55,56,57,58,59, studies revealed lineage relationships in clones derived from diverse classes of progenitor and stem cells (see Table 1 for current list of MADM clonal studies). Another interesting future application of MADM is to combine it with additional functional or subcellular reporters, which would increase the degree of information that can be acquired from clones.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank all members of the Hippenmeyer laboratory for discussion, the Bioimaging Facility, Life Science Facility, and Pre-Clinical Facility at IST Austria for technical support. This work was supported by IST Austria institutional funds; R.B. received support from Austrian Science Fund (FWF) Lise-Meitner program (M 2416); N.A received support from Austrian Science Fund (FWF) Firnberg-Programm (T 1031); GC received support from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 754411 as an ISTplus postdoctoral fellow; A.H. received support from an ÖAW DOC (Doctoral Fellowship of the Austrian Academy of Sciences). This study was also supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 725780 LinPro) to S.H.
Materials
| Name | Company | Catalog Number | Comments |
| 1 mL tuberculin syringe (Omnifix Luer Lock) | Braun | 9204512N | |
| 1,4-diazabicyclooctane (DABCO) | Roth | 0718.2 | |
| 10 mL Syringe (Omnifix Luer Lock) | Braun | 8508429N | |
| 15 mL conical centrifuge | Sarstedt | 65.554.502 | |
| 24 multi-well dishes | Roth/Greiner Bio-one | CE56.1 | |
| 27- gauge x 3/4 needle (Sterican) | Braun | 16010256E | |
| Corn oil | Sigma | C8267-500ML | |
| Coverslips (24 x 60 mm #1) | Thermo Fisher Scientific (Menzel) | 15747592 | |
| Cryostat Cryostar NX70 | Thermo Fisher Scientific | 957000H | |
| Dako Pen (Wax marker) | Agilent | S200230-2 | |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 | |
| Disposable microtome blade (MX35 Ultra) | Thermo Fisher Scientific | 705830 | |
| Fine Forceps (Dumont #5) | Fine Science Tools (FST) | 11254-20 | |
| Glass anti-roll plate | Histocom | M 449980 | |
| Glycerol | Sigma | G5516 | |
| LSM 800 Confocal | Zeiss | ||
| Mounting medium | 25 mg/mL DAPCO, 6 g Glycerol, 2.4 g Mowiol 4-88, 6 mL dH2O, 12 mL 0.2 M Tris-HCl (pH 8.5) | ||
| Mowiol 4-88 | Roth | 0713.2 | |
| Normal donkey serum | Innovative Research | IGDNSER100ML | |
| Paraformaldehyde | Sigma | 441244-1KG | |
| Peristaltic pump 323E/D 400RPM | Watson-Marlow | 036.3124.00A | |
| Sucrose | Sigma | S8501-5KG | |
| Superfrost plus glass slides | Thermo Fisher Scientific | J1800AMNT | |
| Tamoxifen | Sigma | T5648 | |
| Tissue Embedding mold T-12 (22mm square) | Polysciences Inc. | 18986-1 | |
| Tissue-Tek O.C.T | Sakura | 4583 | |
| Triton X-100 | Sigma | T8787-250ML | |
| Trizma hydrochloride | Sigma | 93363 | |
| Tween-20 | Sigma | P9416-100ML | |
| Software and Plugins: | |||
| Fiji | 1.52p | Fiji | |
| MultiStackReg | 1.45 | Download link | |
| TurboReg | EPFL Bioimaging | ||
| Zen Blue | 2.6 | Zeiss | |
| Experimental Models: Organisms/Strains: | |||
| Mouse: Emx1-CreER | The Jackson Laboratory | JAX:027784 | |
| Mouse: MADM-11-GT | The Jackson Laboratory | JAX:013749 | |
| Mouse: MADM-11-TG | The Jackson Laboratory | JAX:013751 | |
| Primary antibodies: | |||
| Chicken anti-GFP 1:500 | Aves Labs | GFP-1020 | |
| Goat anti-tdTomato 1:500 | Sicgen Antibodies | AB8181-200 | |
| Rabbit anti-RFP 1:500 | MBL | PM005 | |
| Secondary antibodies: | |||
| Donkey Anti-Chicken Alexa Fluor 488 1:500 | Jackson Immuno Research | 715-475-150 | |
| Donkey Anti-Goat Cy3 1:500 | Jackson Immuno Research | 705-165-147 | |
| Donkey Anti-Rabbit Cy3 1:500 | Jackson Immuno Research | 711-165-152 |
References
- Malatesta, P., et al. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 37 (5), 751-764 (2003).
- Miyata, T., Kawaguchi, A., Okano, H., Ogawa, M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron. 31 (5), 727-741 (2001).
- Noctor, S. C., Flint, A. C., Weissman, T. A., Dammerman, R. S., Kriegstein, A. R. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 409 (6821), 714-720 (2001).
- Taverna, E., Götz, M., Huttner, W. B. The Cell Biology of Neurogenesis: Toward an Understanding of the Development and Evolution of the Neocortex. Annual Review of Cell and Developmental Biology. 30 (1), 465-502 (2014).
- Desikan, R. S., Barkovich, A. J. Malformations of cortical development. Annals of Neurology. 80 (6), 797-810 (2016).
- Gao, R., Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Current Molecular Medicine. 15 (2), 146-167 (2015).
- Beattie, R., et al. Mosaic Analysis with Double Markers Reveals Distinct Sequential Functions of Lgl1 in Neural Stem Cells. Neuron. 94 (3), 517-533 (2017).
- Hippenmeyer, S. Molecular pathways controlling the sequential steps of cortical projection neuron migration. Advances in Experimental Medicine and Biology. 800, 1-24 (2014).
- Lodato, S., Arlotta, P. Generating neuronal diversity in the mammalian cerebral cortex. Annual Review of Cell and Developmental Biology. 31, 699-720 (2015).
- Hansen, A. H., Duellberg, C., Mieck, C., Loose, M., Hippenmeyer, S. Cell Polarity in Cerebral Cortex Development-Cellular Architecture Shaped by Biochemical Networks. Frontiers in Cellular Neuroscience. 11, 176 (2017).
- Gao, P., et al. Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell. 159 (4), 775-788 (2014).
- Llorca, A., et al. A stochastic framework of neurogenesis underlies the assembly of neocortical cytoarchitecture. eLife. 8, e51381 (2019).
- Ma, J., Shen, Z., Yu, Y. C., Shi, S. H. Neural lineage tracing in the mammalian brain. Current Opinion in Neurobiology. 50, 7-16 (2018).
- Caviness, V., Takahashi, T., Nowakowski, R. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends in Neurosciences. 18 (9), 379-383 (1995).
- Schmechel, D. E., Rakic, P. A Golgi study of radial glial cells in developing monkey telencephalon: morphogenesis and transformation into astrocytes. Anatomy and Embryology. 156 (2), 115-152 (1979).
- Kessaris, N., et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nature Neuroscience. 9 (2), 173-179 (2006).
- Magavi, S., Friedmann, D., Banks, G., Stolfi, A., Lois, C. Coincident generation of pyramidal neurons and protoplasmic astrocytes in neocortical columns. The Journal of Neuroscience. 32 (14), 4762-4772 (2012).
- Anthony, T. E., Klein, C., Fishell, G., Heintz, N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron. 41 (6), 881-890 (2004).
- Voigt, T. Development of glial cells in the cerebral wall of ferrets: direct tracing of their transformation from radial glia into astrocytes. The Journal of Comparative Neurology. 289 (1), 74-88 (1989).
- Amberg, N., Laukoter, S., Hippenmeyer, S. Epigenetic cues modulating the generation of cell-type diversity in the cerebral cortex. Journal of Neurochemistry. 149 (1), 12-26 (2019).
- Beattie, R., Hippenmeyer, S. Mechanisms of Radial Glia Progenitor Cell Lineage Progression. FEBS letters. 591 (24), 3993-4008 (2017).
- Telley, L., et al. Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science. 364 (6440), eaav2522 (2019).
- Oberst, P., et al. Temporal plasticity of apical progenitors in the developing mouse neocortex. Nature. 573 (7774), 370-374 (2019).
- Telley, L., et al. Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science. 351 (6280), 1443 (2016).
- Deppe, U., et al. Cell lineages of the embryo of the nematode Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 75 (1), 376-380 (1978).
- Woodworth, M. B., Girskis, K. M., Walsh, C. A. Building a lineage from single cells: genetic techniques for cell lineage tracking. Nature Reviews Genetics. 18 (4), 230-244 (2017).
- Masuyama, N., Mori, H., Yachie, N. DNA barcodes evolve for high-resolution cell lineage tracing. Current Opinion in Chemical Biology. 52, 63-71 (2019).
- Legue, E., Joyner, A. L. Chapter Ten-Genetic Fate Mapping Using Site-Specific Recombinases. Methods in Enzymology. 477, 153-181 (2010).
- Postiglione, M. P., Hippenmeyer, S. Monitoring neurogenesis in the cerebral cortex: an update. Future Neurology. 9 (3), 323-340 (2014).
- Espinosa-Medina, I., Garcia-Marques, J., Cepko, C., Lee, T. High-throughput dense reconstruction of cell lineages. Open Biology. 9 (12), 190229 (2019).
- Hwang, B., et al. Lineage tracing using a Cas9-deaminase barcoding system targeting endogenous L1 elements. Nature Communications. 10 (1), 1234 (2019).
- Kalhor, R., et al. Developmental barcoding of whole mouse via homing CRISPR. Science. 361 (6405), eaat9804 (2018).
- García-Moreno, F., Vasistha, N. A., Begbie, J., Molnár, Z. CLoNe is a new method to target single progenitors and study their progeny in mouse and chick. Development. 141 (7), 1589-1598 (2014).
- Loulier, K., et al. Multiplex Cell and Lineage Tracking with Combinatorial Labels. Neuron. 81 (3), 505-520 (2014).
- Livet, J., et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 450 (7166), 56-62 (2007).
- Snippert, H. J., et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell. 143 (1), 134-144 (2010).
- Amitai-Lange, A., et al. A method for lineage tracing of corneal cells using multi-color fluorescent reporter mice. Journal of Visualized Experiments. (106), e53370 (2015).
- Vasistha, N. A., et al. Cortical and Clonal Contribution of Tbr2 Expressing Progenitors in the Developing Mouse Brain. Cerebral Cortex. 25 (10), 3290-3302 (2015).
- Chen, F., LoTurco, J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. Journal of Neuroscience Methods. 207 (2), 172-180 (2012).
- Siddiqi, F., et al. Fate mapping by piggyBac transposase reveals that neocortical GLAST+ progenitors generate more astrocytes than Nestin+ progenitors in rat neocortex. Cerebral Cortex. 24 (2), 508-520 (2014).
- Yoshida, A., et al. Simultaneous expression of different transgenes in neurons and glia by combining in utero electroporation with the Tol2 transposon-mediated gene transfer system. Genes to Cells. 15 (5), 501-512 (2010).
- García-Marqués, J., López-Mascaraque, L. Clonal Identity Determines Astrocyte Cortical Heterogeneity. Cerebral Cortex. 23 (6), 1463-1472 (2013).
- Clavreul, S., et al. Cortical astrocytes develop in a plastic manner at both clonal and cellular levels. Nature Communications. 10 (1), 4884 (2019).
- Kim, G. B., et al. Rapid Generation of Somatic Mouse Mosaics with Locus-Specific, Stably Integrated Transgenic Elements. Cell. 179 (1), 251-267 (2019).
- Pilz, G. A., et al. Live imaging of neurogenesis in the adult mouse hippocampus. Science. 359 (6376), 658 (2018).
- Hippenmeyer, S., et al. Genetic Mosaic Dissection of Lis1 and Ndel1 in Neuronal Migration. Neuron. 68 (4), 695-709 (2010).
- Zong, H., Espinosa, J. S., Su, H. H., Muzumdar, M. D., Luo, L. Mosaic analysis with double markers in mice. Cell. 121 (3), 479-492 (2005).
- Ramón y Cajal, S. . Histologie du système nerveux de l'homme et des vertébrés. , (1911).
- Cowan, W. M. The emergence of modern neuroanatomy and developmental neurobiology. Neuron. 20 (3), 413-426 (1998).
- Ali, S. R., et al. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proceedings of the National Academy of Sciences of the United States of America. 111 (24), 8850-8855 (2014).
- Hippenmeyer, S., Johnson, R. L., Luo, L. Mosaic analysis with double markers reveals cell-type-specific paternal growth dominance. Cell Reports. 3 (3), 960-967 (2013).
- Riccio, P., Cebrian, C., Zong, H., Hippenmeyer, S., Costantini, F. Ret and Etv4 promote directed movements of progenitor cells during renal branching morphogenesis. PLoS Biology. 14 (2), e1002382 (2016).
- Bonaguidi, M. A., et al. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. 145 (7), 1142-1155 (2011).
- Mayer, C., et al. Clonally Related Forebrain Interneurons Disperse Broadly across Both Functional Areas and Structural Boundaries. Neuron. 87 (5), 989-998 (2015).
- Muzumdar, M. D., et al. Clonal dynamics following p53 loss of heterozygosity in Kras-driven cancers. Nature Communications. 7, 12685 (2016).
- Shi, W., et al. Ontogenetic establishment of order-specific nuclear organization in the mammalian thalamus. Nature Neuroscience. 20, 516 (2017).
- Wong, S. Z. H., et al. In vivo clonal analysis reveals spatiotemporal regulation of thalamic nucleogenesis. PLoS Biology. 16 (4), e2005211 (2018).
- Xu, H. T., et al. Distinct Lineage-Dependent Structural and Functional Organization of the Hippocampus. Cell. 157 (7), 1552-1564 (2014).
- Brennand, K., Huangfu, D., Melton, D. All β Cells Contribute Equally to Islet Growth and Maintenance. PLoS Biology. 5 (7), e163 (2007).
- Ortiz-Alvarez, G., et al. Adult neural stem cells and multiciliated ependymal cells share a common lineage regulated by the geminin family members. Neuron. 102 (1), 159-172 (2019).
- Kaplan, E. S., Ramos-Laguna, K. A., Mihalas, A. B., Daza, R. A. M., Hevner, R. F. Neocortical Sox9+ radial glia generate glutamatergic neurons for all layers, but lack discernible evidence of early laminar fate restriction. Neural Development. 12 (1), 14 (2017).
- Lv, X., et al. TBR2 coordinates neurogenesis expansion and precise microcircuit organization via Protocadherin 19 in the mammalian cortex. Nature Communications. 10 (1), 3946 (2019).
- Mihalas, A. B., Hevner, R. F. Clonal analysis reveals laminar fate multipotency and daughter cell apoptosis of mouse cortical intermediate progenitors. Development. 145 (17), dev164335 (2018).
- Picco, N., et al. A mathematical insight into cell labelling experiments for clonal analysis. Journal of Anatomy. 235 (3), 687-696 (2019).
- Johnson, C. A., Ghashghaei, H. T. Sp2 regulates late neurogenic but not early expansive divisions of neural stem cells underlying population growth in the mouse cortex. Development. , (2020).
- Espinosa, J. S., Luo, L. Timing Neurogenesis and Differentiation: Insights from Quantitative Clonal Analyses of Cerebellar Granule Cells. The Journal of Neuroscience. 28 (10), 2301 (2008).
- Schneider, C. A., Rasband, W. S., Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 9 (7), 671-675 (2012).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Liu, Y., et al. Tamoxifen-independent recombination in the RIP-CreER mouse. PLoS One. 5 (10), e13533 (2010).
- Klingler, E., Jabaudon, D. Do progenitors play dice?. eLife. 9, e54042 (2020).
- Hippenmeyer, S. Dissection of gene function at clonal level using mosaic analysis with double markers. Frontiers in Biology. 8 (6), 557-568 (2013).
- Rulands, S., Simons, B. D. Tracing cellular dynamics in tissue development, maintenance and disease. Current Opinion in Cell Biology. 43, 38-45 (2016).
- Laukoter, S., et al. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nature Communications. 11 (1), 195 (2020).
- Daigle, T. L., et al. A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell. 174 (2), 465-480 (2018).
- He, M., et al. Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron. 91 (6), 1228-1243 (2016).
- Yamamoto, M., et al. A multifunctional reporter mouse line for Cre- and FLP-dependent lineage analysis. Genesis. 47 (2), 107-114 (2009).
- Plummer, N. W., et al. Expanding the power of recombinase-based labeling to uncover cellular diversity. Development. 142 (24), 4385 (2015).
- Imayoshi, I., Ohtsuka, T., Metzger, D., Chambon, P., Kageyama, R. Temporal regulation of Cre recombinase activity in neural stem cells. Genesis. 44 (5), 233-238 (2006).
- Sasaki, S., et al. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Molecular and Cellular Biology. 25 (17), 7812-7827 (2005).
- Acampora, D., et al. Epilepsy and brain abnormalities in mice lacking the Otx1 gene. Nature Genetics. 14 (2), 218-222 (1996).
- Soeda, T., et al. Sox9-expressing precursors are the cellular origin of the cruciate ligament of the knee joint and the limb tendons. Genesis. 48 (11), 635-644 (2010).
- Klezovitch, O., Fernandez, T. E., Tapscott, S. J., Vasioukhin, V. Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes & Development. 18 (5), 559-571 (2004).
- Pimeisl, I. M., et al. Generation and characterization of a tamoxifen-inducible EomesCreER mouse line. Genesis. 51 (10), 725-733 (2013).
- Nakagawa, N., et al. Memo1-Mediated Tiling of Radial Glial Cells Facilitates Cerebral Cortical Development. Neuron. 103 (5), 836-852 (2019).
- Nowotschin, S., et al. The T-box transcription factor Eomesodermin is essential for AVE induction in the mouse embryo. Genes & Development. 27 (9), 997-1002 (2013).
- Balordi, F., Fishell, G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. Journal of Neuroscience. 27 (52), 14248-14259 (2007).
- Liang, H., et al. Neural development is dependent on the function of specificity protein 2 in cell cycle progression. Development. 140 (3), 552-561 (2013).
- Guo, C., Yang, W., Lobe, C. G. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis. 32 (1), 8-18 (2002).
- Ahn, S., Joyner, A. L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 437 (7060), 894-897 (2005).
- Storm, R., et al. The bHLH transcription factor Olig3 marks the dorsal neuroepithelium of the hindbrain and is essential for the development of brainstem nuclei. Development. 136 (2), 295-305 (2009).
- Koundakjian, E. J., Appler, J. L., Goodrich, L. V. Auditory neurons make stereotyped wiring decisions before maturation of their targets. Journal of Neuroscience. 27 (51), 14078-14088 (2007).
- Dor, Y., Brown, J., Martinez, O. I., Melton, D. A. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 429 (6987), 41-46 (2004).
- Sohal, D. S., et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circulation Research. 89 (1), 20-25 (2001).
- Ventura, A., et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 445 (7128), 661-665 (2007).
- Johnson, L., et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 410 (6832), 1111-1116 (2001).
- Tasic, B., et al. Extensions of MADM (mosaic analysis with double markers) in mice. PLoS One. 7 (3), e33332 (2012).
- Yu, J., Carroll, T. J., McMahon, A. P. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development. 129 (22), 5301-5312 (2002).
- Zhao, H., et al. Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Developmental Biology. 276 (2), 403-415 (2004).
- Schuchardt, A., D'Agati, V., Larsson-Blomberg, L., Costantini, F., Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 367 (6461), 380-383 (1994).
- Livet, J., et al. ETS gene Pea3 controls the central position and terminal arborization of specific motor neuron pools. Neuron. 35 (5), 877-892 (2002).
- Volz, K. S., et al. Pericytes are progenitors for coronary artery smooth muscle. eLife. 4, e10036 (2015).
- Cai, C. L., et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 454 (7200), 104-108 (2008).
- Lasrado, R., et al. Lineage-dependent spatial and functional organization of the mammalian enteric nervous system. Science. 356 (6339), 722-726 (2017).
- Matsuoka, T., et al. Neural crest origins of the neck and shoulder. Nature. 436 (7049), 347-355 (2005).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved